
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Loading...
Accelerate your genomics journey with Illumina Connected Software. Unlock insights, streamline workflows, and collaborate seamlessly—all in one integrated ecosystem.
Illumina Connected Software refers to Illumina's portfolio of software products, including BaseSpace Sequence Hub, Illumina Connected Analytics, and more.
The following steps represent the end-to-end user journey, from managing workflows in the lab to interpreting data from analyses. Click on each step in the workflow for relevant support provided by Illumina's suite of software solutions.
Log in to your Illumina Connected Software account . You can also click "Sign In" in the top right. Please note that an account is required to log in. To register your software and add users, refer to the instructions provided in the section.
Need help getting started? Explore our resources to learn more about software registration, administration options, single sign-on and more.
Discover comprehensive information across various software products to streamline your workflow, including , , , , and . Supplement your learning with designed to enhance your proficiency and usage.
Explore, learn, and connect with the tools you need, all in one place.
You can search our help documentation or ask questions with AI-generated answers using the search-box at the top of the page.
Navigate and explore using the left-panel.
Assay design for arrays and sequencing
Sample and workflow management
Sequencing and analysis set up
Variant calling, data management and beyond
Interpretation and reporting
Sample collection to multiomic insight





Connected Home's Applications Page is the landing central hub for managing account in Illumina.
Note: "Connected Home" and "Platform Home" terms are used interchangeably.
Once you have , you can log in through the to access Connected Home. From this page, you can click on any app tile to launch a product.
Once you're inside an application, you can return to Connected Home at any time by clicking on the waffle menu present to the top right corner and selecting "Platform Home." From the waffle menu, you can also switch between any of the applications you have access to.
The left navigation menu in Connected Home provides various options to manage account in Illumina.
The Illumina Usage Explorer allows you to track your usage costs across Illumina cloud products with detailed insights and trends. To learn more about the usage explorer visit .
In the left navigation menu, the User component features a dropdown list containing options for managing the following:
Profile
API keys
Sessions
Passwords
The "Profile" option takes you to a page where you can update your profile information. Please note that the email address associated with your profile cannot be changed. However, you can edit the following fields: first name, last name, company name, job title, and country/region.
Click "Save" to apply your changes, or "Cancel" to discard them.
The "API Keys" page allows you to view and manage all your API keys.
Manage Existing API Keys
You will see a list of all the API keys associated with your domain, including their status, creation date, expiration date, last used date, and available actions. You can expand the dropdown for each key to view the roles associated with it, if applicable.
Click the icon to generate a new version of an existing API key. Regenerating an API key creates a new key value that replaces the old one. This is often done for security reasons—such as when you suspect a key has been exposed—or to rotate credentials regularly. Please note: if you regenerate a key, any existing integrations that rely on the original key may stop working unless you update them with the new key.
Click the icon to edit an API key. You can update the key name and choose whether the changes apply to all future workgroups and roles within the domain. You can also specify which current workgroups and domains the updated name applies to.
Click theicon to delete an API key. You can no longer access the API key once it's deleted.
Generate New API Key
To create a new API key, click the "Generate" button. Provide a name for the key, then choose to either include all workgroups or select specific workgroups that the key should have access to.
Once generated, the API key will be displayed in a hidden format. Use the "Show Key" button to reveal it, and optionally download the key as a file for secure storage.
⚠️ Important: Once this window is closed, the key will no longer be accessible through the domain interface. Be sure to store it securely for future use.
After generating a key, keep it in a safe location to use when accessing the platform via the command-line interface (CLI) or APIs.
The "Sessions" page takes you to a table displaying a log of your user account session activities. Activities such as sign-ins, session generations, and sign-outs from different devices are recorded.
You’ll also see additional columns providing details about each activity, including the device, browser, IP address, creation time, last activity time, and action.
From the Action column, you can click "Sign Out" to end any active sessions.
Lastly, click the waffle menu next to each column header to pin, autosize, reset, or customize the ccolumns displayed.
The Password page allows you to change the password for the domain.
The Communication page allows you to choose the communication preference from Illumina
The "Subscriptions" link takes you to a table displaying all subscriptions associated with the domain. This includes details such as the subscription name, subscription number, status, start date, end date, email address, and available actions.
The end date indicates when the subscription is scheduled to expire.
Most subscriptions—such as ICA—provide access to applications for all users in a domain.
For certain subscriptions—such as —specific users need to be assigned to the subscription in order for someone to access the associated application. Only domain admins have permission to assign or unassign users to these type of subscriptions. To assign a user, click "Assign" and enter the user’s email address.
To unassign a user, click the "Unassign" link and the user will be removed.
Click the waffle menu next to each column to filter, pin, autosize, reset, or customize the columns displayed.
The Orders Registration page allows you to set up and manage software orders. To learn more about the Order Registration and Software setup visit .
The Illumina Admin console is a platform designed for administrators to manage product usage, control workgroup permissions, and oversee domain access. To learn more about the Roles, Workgroup and Domain visit .
Custom design software helps create custom microarray probes and sequencing panels optimized for specific genomic content of interest. These software tools support a broad range of applications, including custom microarray-based genotyping applications, custom targeted amplicon sequencing, and custom target enrichment.
Refer to the respective documentation sites via the links below for more information. Access the full list of Illumina software products at the .
The sample sheet for each instrument contains fields that must be filled accurately to specify the analyses to be performed. Refer to the instrument settings for sample sheet instructions specific to your instrument.
Communications
















Run set up refers to configuring settings for sequencing on Illumina instruments. In addition, users can provide settings for data analysis, location of the output data, and other parameters.
Sequencing runs can be set up using software applications and by downloading and editing sample sheet templates that are uploaded into the sequencer. The Run Planning tool in BaseSpace Sequence Hub and sample sheets are both generic and support run set-up for a broad spectrum of instruments and analysis pipelines. Other software applications support run set-up and analysis only for specific instruments, assays, and analysis pipelines.
Refer to the respective documentation sites via the links below for more information. Access the full list of Illumina software products at the Illumina Support Center.
The Illumina Admin Console is a platform designed for administrators to manage product usage, control workgroup permissions, and oversee domain access.
To access the Admin Console, log in through the , to go to the . From there, select "Admin" from the left navigation menu. You can also access it directly at https://<domain>.login.illumina.com/iam.
A sample sheet is a comma-separated value (*.csv) file format used by Illumina instruments, platforms, and analysis pipelines to store settings and data for sequencing and analysis. The sample sheet uses American Standard Code for Information Interchange (ASCII) character encoding.
The system using the sample sheet as input validates the data within the sample sheet based on validation rules. Validation rules differ from section to section. Incorrectly formatted data are considered invalid and causes software to throw an error. Refer to the validation rules for each section for more information on acceptable formatting and allowable values.
Please see the pages below for troubleshooting guidance.
Lab management software enables labs to track samples, streamline complex tasks, generate sample sheets, and catch poor quality samples before running them on a sequencing system.
Refer to the respective documentation sites via the links below for more information. Access the full list of Illumina software products at the .








🗓️ Includes Run Planning tool for run set-up and sample sheet generation.
🗒️ Customizable templates for configuring sequencing runs and analysis settings.


IAM = Identity and Access Management
IAM Console and Admin Console are used interchangeably.
The roles within a domain that users may be assigned are:
Domain Admin
Read/write access to all resources created by users in the domain. Manage domain and workgroup membership.
Workgroup Admin
Read/write access to all resources created by users in the workgroup. Manage workgroup membership.
Workgroup User
Read/write access to all resources created by users in the workgroup.
Click on each section below to explore the different features of the Admin Console.

A v2 sample sheet can be obtained from one of the following options.
A v2 sample sheet can be created in BaseSpace using Run Planning.
In BaseSpace Sequence Hub, select the Runs tab > New Run > Run Planning > Local Mode > Export Sample Sheet.
For this option, all sample and index information is entered in Run Planning and is included in the exported v2 sample sheet.
A v2 sample sheet template can be downloaded and filled out manually.
Download the template with the link below and use a text editor program to alter the sections according to your chosen instrument or analysis. Using a spreadsheet editor can cause errors with sample sheet validation and analysis. Save the sample sheet to a folder accessible by your instrument or analysis software.
Follow instructions to fill out the sample sheet. See resources below:
Download
Frequently Asked Questions
A: There are two types of software that use the registration process to activate:
Software Subscriptions
BaseSpace Professional Subscriptions
ICA Professional, ICA Enterprise, Illumina Connected Insights, Illumina Connected Multiomics Subscriptions, etc.
Software Consumables
Examples: iCredits, Genome Equivalent Samples, Illumina Complete Long Reads cloud analysis, Consumption billing
A: Workgroups and Domains are used by some Illumina Software to control access to different customer’s data and assets. Examples of a domain are dcehealth.Illumina.com or coreseqinc.Illumina.com.
Choosing a domain is required when registering BaseSpace Enterprise, ICA, or Cohorts Subscriptions. Choosing a workgroup is required when registering BaseSpace Professional. Software consumables (such as iCredits) can be applied to either domains or workgroups of active subscriptions.
Below is a description of how workgroups and domains are used by Illumina Software.
A: If you arrive at the Software Registration Portal page and see no orders listed, this means that the orders have not been associated with your email address.\
You have most likely been forwarded an Illumina registration email and clicked on the link to get to the registration portal. The portal does not recognize your email because the person that forwarded the message did not request that your email be assigned to register new products.
The solution for this is to ask the person who forwarded the email to you to log into the registration portal and add your email address using the “Assign User” button. If this does not resolve the issue or you are unable to locate your order after logging in, please contact [email protected] or click on the link on the Registration Page.
A: After registration is completed, a notification will be sent to your email address to confirm the account details.
Provisioning of your software in the cloud can take up to 15-30 minutes after registration is complete. When provisioning is completed, you will receive a welcome notification that includes getting started information about your subscription.
The information contained within a sample sheet is organized into various sections, like Header, Reads, Manifests, Data, and Settings. There are two types of sections:
Standalone sections—Contain run settings and data that do not pertain to a specific application.
Applications sections—Contain run settings and data required by specific applications.
The Header and Reads sections contain run settings and data. The Header and Reads sections are present in every v2 sample sheet. Additional standalone sections such as Sequencing Settings can be added and do not produce an error.
A new section includes a section header that precedes the data contained within the section. A section header consists of the name of the section within brackets, as seen in the first line of the following example.
The application sections contain data required to run Illumina applications. Application settings differ from instrument to instrument. Refer to the applicable section.
Designate an applications section by appending Settings or Data to the section name as seen in the following example.
Each application can have a maximum of one Settings section and one Data section.
Settings section—This section contains key-value pairs that configure settings for the application, as seen in the example. The Settings section is applied globally to all samples.
Data section—This section contains per sample data (in tabular format) used as input by the given application. The data in the first non-empty line of the application data section forms the column headers for the rest of the section. The Data section is uniquely applied to each sample.
The Cloud Orchestrated Cloud Analysis workflow streamlines sequencing run planning and cloud-based secondary analysis using the BaseSpace Sequence Hub Run Planning interface. With Cloud Orchestrated Cloud Analysis, you can plan a sequencing run for a specific instrument platform and configure the associated cloud secondary analysis using the BaseSpace Sequence Hub Run Planning interface. Once the run has been planned, any instrument of that platform can access and kick off the run, provided you log into the instrument using the same workgroup context as the run was planned in.
Sequencing runs using this workflow require you to upload your run data to BaseSpace Sequence Hub so that it can be used to run the associated secondary analysis.
Once the sequencing run is complete and all primary analysis data has been uploaded to BaseSpace Sequence Hub, the secondary analysis is automatically kicked off in the cloud and linked to the run in BaseSpace Sequence Hub.
Current instrument platforms that support Cloud Orchestrated Cloud Analysis:
NextSeq 1000 / NextSeq 2000
NovaSeq X Series
The Cloud Orchestrated Local Analysis workflow enables you to plan your sequencing run (associated with a specific instrument platform) and the associated on-instrument secondary analysis using the BaseSpace Sequence Hub Run Planning interface. Once the run has been planned, any instrument of that platform can access and kick off the run, provided you log into the instrument using the same workgroup context as the run was planned in.
Sequencing runs using the Cloud Orchestrated Local Analysis workflow require you to send at least run monitoring data to BaseSpace Sequence Hub so that the run planned in the cloud can be kept in sync with the instrument.
Once the sequencing run is complete and all primary analysis data has been generated, the secondary analysis is automatically kicked off on the instrument as specified by the user during run planning.
Current instrument platforms that support Cloud Orchestrated Local Analysis:
NextSeq 1000 / NextSeq 2000
MiSeq i100 Series
The Locally Orchestrated Local Analysis workflow enables you to plan a sequencing run and associated on-instrument secondary analysis using the on-instrument Run Planning interface. Once the run has been planned, any user can access and kick off the run upon logging into the instrument.
Once the sequencing run is complete and all primary analysis data has been generated, the secondary analysis is automatically launched on the instrument, as specified by the user during run planning.
Sequencing runs using the Locally Orchestrated Local Analysis workflow enable you to use any level of cloud connectivity and data upload. On NovaSeq X Series or MiSeq i100 Series instruments, you can also optionally upload FastQ and BAM/CRAM files to the cloud if desired.
Current instrument platforms that support Locally Orchestrated Local Analysis:
NovaSeq X Series
MiSeq i100 Series
The Sample Sheet Driven Local Analysis workflow leverages DRAGEN on the instrument, relying on a sample sheet to provide the necessary details for orchestrating and executing the analysis directly onboard. In order to use this workflow, select the option when setting up a run to perform local analysis using a sample sheet, and attach the desired sample sheet to your run.
When using this workflow, the DRAGEN analysis is started onboard automatically when the sequencing data is in a ready state to begin analysis.
For optimal results, it is recommended to generate the initial sample sheet using the BaseSpace Sequence Hub Run Planning interface for local analysis. This will ensure proper formatting and acceptable values for all fields.
Current instrument platforms that support Sample Sheet Driven Local Analysis:
NextSeq 1000 / NextSeq 2000
NovaSeqX Series
The Illumina Usage Explorer allows you to track your usage costs across Illumina cloud products with detailed insights and trends.
To access the Usage Explorer, log in through the domain login URL, and navigate to the product dashboard. From there, select "Usage Explorer" from the list of available applications.
The Usage Explorer can also be accessed by clicking the top left waffle menu and selecting "Usage Explorer".
The summary section gives you a quick look at your iCredit balance, iCredit usage for the current month, quarter, and year, and total storage separated by tier. If you have purchased or used cost units other than iCredits (e.g., GEs, Samples, etc), they will be displayed under additional tabs.
Domain admins can set a budget threshold, and if set it will display in the summary section. To set a budget threshold, switch on the budget notifications toggle and click "Edit budget settings" to specify an iCredit threshold and any email recipients.
The Trend & Details section gives you the ability to filter, group, and explore your usage. The controls on the right side have a variety of options that allow you to examine your data at the granularity you choose. Above the chart, usage and storage metrics are displayed that update with the filter settings.
Below the chart, a table summarizes the data and can be exported as a CSV file. The usage shown depends on several factors including your subscription and admin status. For example, a domain admin has access to all domain usage, including users, workgroups, and ICA projects, while a workgroup or ICA project admin has access to usage within that specific workgroup or project, respectively.
The reports tab allows you to download a CSV-formatted report of your usage costs for a given time period. Use the date choosers to select a range for the report, up to three months at a time.
By default, the reports are generated in the latest usage report format. This newer format is more detailed, but the legacy option is also available to preserve backwards compatibility.
Multiomics software offers a comprehensive range of solutions to streamline your journey from sample collection to multiomic insights. Discover analysis for designed for protein data, spatial data, single-cell RNA data, and more.
Our powerful study management tool helps organize your data from ingestion to analysis, while our advanced analysis platform generates actionable insights to drive your research forward.
Refer to the respective documentation sites via the links below for more information. Access the full list of Illumina software products at the .
Interpretation software includes products that help generate meaningful clinical insights from NGS and other data. They cover statistical and visualization tools, solutions for cohort analysis, variant interpretation, and report generation.
Refer to the respective documentation sites via the links below for more information. Access the full list of Illumina software products at the .
Data analysis software includes a broad spectrum of products covering variant calling, annotation, pipeline development, data management, storage and sharing. The products cover multiple applications (eg, germline, somatic, infectious disorders, and microbiology), data types (eg, sequencing, microarrays, multiomics), analysis throughput, deployment mode (eg, cloud, on-premises, on-board), and other options.
Refer to the respective documentation sites via the links below for more information. Access the full list of Illumina software products at the .
MiSeq i100 Series
MiSeq i100 Series
BaseSpace Sequence Hub Professional
Workgroup
BaseSpace Pro subscriptions use workgroups to control access to software consumables.
All other software products (ICA, ICI, ICM, etc.)
Domain
All other software subscriptions use domains to control access to software consumables.

Manage workgroup settings and user roles
Manage domain settings










[Header],,,
FileFormatVersion,2,,
RunName,MyRun,,
InstrumentPlatform,NextSeq1k2k,,
InstrumentType,NextSeq2000,,
,,,<Enter analysis settings for demultiplexing. Used by BCL Convert software.>,,,
[BCLConvert_Settings],,,
SoftwareVersion,x.y.z,,
,,,
<Enter sample information of each sample in the run for demultiplexing. Used by BCL Convert software.>,,,
[BCLConvert_Data],,,
Lane,Sample_ID,index,index2
1,S01-TOO-12plex-P1-rep1,ATCCACTG,AGGTGCGT
1,S02-TOO-12plex-P1-rep2,GCTTGTCA,GAACATAC
1,S03-TOO-12plex-P1-rep3,GCTTGTCA,GAACATAC
1,S04-TOO-12plex-P1-rep4,GCTTGTCA,GAACATACSelect the BaseSpace tab.
Optional: Select Turn on Illumina Proactive Support to enable the Illumina Proactive monitoring service.
Select the region the instrument should connect to. This setting is used to ensure proper data storage is sent to Illumina Proactive Support and BaseSpace Sequence Hub.
Optional: Select Private Domain and enter the private domain of your Enterprise BaseSpace Sequence Hub account, if applicable.
From the Home screen, select Sequence.
From the Run Setup Option screen, select Manual.
From the BaseSpace Options screen, select Use BaseSpace™ Sequencing Hub for this run.
Select the Run analysis, collaboration, and storage option to send all run data to BaseSpace Sequence Hub.
Enter your BaseSpace Sequence Hub account credentials.
Select Sign In.
Select the Workgroup.
Select Next.
Set the Read Type, Read Length and Custom Primers for all of your cycles.
Browse to select your sample sheet file. See for information on how to download a sample sheet file.
Select Next.
Continue to set up the sequencing run as per the .
When performing dual indexing, all Illumina sequencers read the first index (i7) in the same direction; however, the second index (i5) is read in different orientations depending on the instrument and chemistry, in particular, differences between when the paired-end turnaround and i5 read take place:
On the MiSeq, HiSeq 2000/2500, and the MiniSeq Rapid kits the i5 sequence is read in the forward orientation.
On the iSeq 100, MiniSeq, NextSeq 500/500, NextSeq 1000/2000, HiSeq 3000/4000/X systems, NovaSeq 6000 (v1.5 reagents), and the NovaSeq X/X Plus the i5 sequence is read in the reverse complement orientation.
See the for information on when to use which orientation of the i5 sequence for these platforms depending on the analysis software used.
When demultiplexing, the final sample sheet provided to the FASTQ generation software needs to have the i5 index in the correct orientation for that platform. However, most Illumina sample sheet generation software handles this automatically. This means that these software work on the assumption that your i5 sequence is always in the forward orientation and will automatically create the reverse complement of the sequences provided for the appropriate instrument type.
The following provides guidance on when to reverse complement and when to not reverse complement the i5 sequences when analyzing iSeq, MiniSeq, NextSeq 500/550, NextSeq 1000/2000, HiSeq 3000/4000/X, NovaSeq 6000 (v1.5 reagents), or NovaSeq X/X Plus data.
For Run Planning in BaseSpace, the output is always a v2 sample sheet for BCL Convert:
NextSeq 1000/2000 and NovaSeq X/X Plus i5 indexes will correctly be output in the forward orientation for later downstream processing. Custom kits should also have the i5 indexes entered in the forward orientation.
The MiniSeq Rapid made a recipe change that uses the grafted oligo for the i5 index priming, so it is read in the forward orientation similar to a MiSeq.
If running in manual mode and using bcl2fastq or BCL Convert, or uploading the sample sheet to BaseSpace as a manual mode run and attaching a sample sheet, enter the i5 sequence in the forward direction. Runs uploaded in manual mode have their data directly passed to bcl2fastq with no further processing.
For Local Run Manager (LRM), input the sequences in the reverse complement when setting up the run so the software automatically creates the forward orientation as needed for analysis. This is the opposite of most of our guidance and is only for the MiniSeq Rapid kits.
When assessing index sets for color balance, the sequence observed by the instrument must be assessed, regardless of the orientation used for the sample sheet. For the i7 index, always asses color balance using the "i7 bases for sample sheet" column orientation. For the i5 index, assess color balance using the forward orientation for MiSeq runs and MiniSeq runs with rapid reagents, and the reverse-complement orientation for all other instruments.
For The NextSeq 1000/2000 and NovaSeq X/X Plus platforms, the RunInfo.xml file includes the IsReverseComplement flag. With this flag, the index sequences that BCL Convert recognizes and outputs are in the following orientations in the Top_Unknown_Barcodes output file.
Index 1: the i7 index column is in the forward orientation (match the orientation if the i7 indexes in the sample sheet).
Index 2: the i5 index column is in the reverse complement orientation (are the reverse complement of the i5 indexes in the sample sheet).
For all instruments, the indexes shown in the Top_Unknown_Barcodes are the sequences as they were sequenced/read by the sequencer.
All sections
Data items cannot contain commas.
Supported line endings are \n, \r\n, and .
Required characters are all ASCII characters except for * and the control characters CR
Standalone
Only key-value pairs are supported.
Each key must have exactly one value.
The same key cannot be specified more than one time in a single standalone section.
Header
There can only be one data item in a section header line.
Section headers are required to start with an opening square bracket, and end with a closing square bracket
Opening and closing brackets are both required, and must be the first and last character in the section header line (other than whitespace and commas)
Application
The application name cannot include Settings or Data.
The application name cannot be empty.
The application name cannot include -.
Application Settings
Settings sections can support only key-value pairs.
Each key can have only one value.
The application name cannot include -.
This table describes how to enter the i5 sequence, Forward (F) or Reverse Complement (RC), depending on the instrument and software used.
LRM: Local Run Manager
IEM: Illumina Experiment Manager
BSSH: BaseSpace Sequence Hub
IRM: Illumina Run Manager
Manual Edit: When manually preparing the Sample Sheet in the text editor
Workgroups are groups of users that can share projects and data. You can view an overview of the workgroups in your domain by clicking on the "Workgroups" tab. For each workgroup, you will see the following information:
Collaboration: Indicates whether your workgroup allows collaboration with users outside the domain.
In this section, we highlight various desired outcomes and provide examples to show how they can be achieved.
For more information on how to use each parameter, see the guide.
LFA standalone section cannot be empty.
Data in standalone sections must not contain brackets.
Each application can have a maximum of one Settings section and one Data section.
Keys and values cannot contain [ or ].
Note: The Settings section is applied globally to all samples.
Application Data
For each line that follows the column header line in the application data section, the number of empty plus non-empty data items cannot be less than the number of column headers.
For each line that follows the column header line in the Application Data section, the number of non-empty values cannot be greater than the number of column headers.
Column headers must all be unique.
Each application data section must contain a column header called Sample_ID.
Column headers and values cannot contain [ or ].
Note: The Data section is uniquely applied to each sample.
NextSeq 500/550
F (LRM, Preptab, IEM)
RC
RC
NextSeq 1000/2000
F (Manual Edit, BSSH Run Planning)
RC
F
NovaSeq 6000 (v1.5 kits)
RC (Manual Edit), F (BSSH Run Planning)
RC
RC
NovaSeq X/X Plus
F (IRM, BSSH Run Planning)
RC
F
MiSeq i100 Series
F (IRM, BSSH Run Planning)
F (index-first) RC (read-first)
F
Instrument
i5 Index Orientation Used Based on Run Setup Tool
Standalone bcl2fastq or BSSH FASTQ Generation
Standalone BCL Convert
MiSeq
F (LRM, IEM)
F
F
MiniSeq
F (LRM, Preptab, IEM)
RC
RC
iSeq 100
F (LRM, IEM)
RC
RC
For other supported platforms (such as the NovaSeq 6000), Run Planning will add a line to the Header section of the v2 sample sheet (IndexOrientation,Forward) and the i5 will be written in the forward orientation and processed correctly downstream.
If generating v2 sample sheets for instruments other than the ones listed, confirm the final sample sheet has the i5 index in the correct orientation as described above.
For runs uploaded manually (i.e., an Illumina tool is not being used to generate the sample sheet), the index orientation depends on which instrument you are using. Please see the Index Orientation Table for information on how to enter the i5 sequence depending on the instrument and software used.
When using IEM, i5 sequences must be entered into these software in the forward orientation in all circumstances and for all instrument types.
The sequences displayed are what is entered into the final sample sheet. Kit definitions have the i5 in the forward orientation. The i5 sequences will be appropriately reverse complemented and displayed in the IEM interface based on instrument/reagent type. IEM will use the instrument/reagent type selected in the setup wizard to generate the final sample sheet with the i5 sequences in the correct orientation.
When using BaseSpace Sequence Hub PrepTab, i5 sequences must be entered into these software in the forward orientation in all circumstances and for all instrument types.
The software automatically creates the reverse complement as needed for analysis. If the sequence is reverse complemented, this will only be shown in the SampleSheetUsed.csv file.
When using LRM, i5 sequences must be entered into these software in the forward orientation in all circumstances and for all instrument types.
The software automatically creates the reverse complement as needed for analysis. If the sequence is reverse complemented, this will only be shown in the SampleSheetUsed.csv file or the SampleSheet.csv file created by LRM at the analysis start. LRM will detect the instrument type from the run files and create a final sample sheet with the i5 indexes in the correct orientation for that platform.
Description: A description of the workgroup.
Owner: The owner of the workgroup.
Created On: The date the workgroup was created.
Active Workgroup: Selected workgroups are in use. Deselecting a workgroup will permanently delete it, and this action cannot be undone.
Workgroups can be created by domain administrators. To create a new workgroup, click the +New button.
Provide a workgroup name, description, and administrator email. Optionally choose to enable collaborators outside of the domain to add users from other domains to the workgroup. This setting cannot be changed after the workgroup has been created.
To view the details of a workgroup, click on the workgroup name. This will redirect you to a screen with additional details outlined below.
From the left-hand navigation menu, you can navigate to the following tabs to view and edit details about your workgroup:
Admins: Manage administrators, owners, and description of a workgroup
Users: Manage existing users and invite new users to a workgroup
Pending Invites: Manage all pending invitations to users of this workgroup
Applications: Manage user access to applications associated with this workgroup
In the Admins tab, you will see an Overview screen displaying details of your workgroup.
You can update the name, description, and owner of the workgroup by clicking "Change Settings." The workgroup owner will automatically be added as a workgroup administrator and user if not already. They are the primary administrator and main point of contact for users within the workgroup.
In the Administrators section, the names and usernames of the workgroup admins will be listed. The roles of each administrator are also listed below each name.
To invite a new admin, click the "Invite" button on the right-hand side and enter the email address of the user you want to add. If you would like to add multiple administrators at once, you can specify multiple email addresses with a comma-separated list. Note that the email format must match the Allowed Emails specified in the Domain tab.
Remove Admin
To remove an admin, select the checkbox next to their name and click the "Remove" button on the right-hand side. A popup will appear to confirm whether you want to remove the admin. Leave the box checked to remove the user from all roles in the workgroup. Uncheck the box if you want to keep the user in the workgroup but remove only their admin permissions.
The Users tab will display the total number of users in the workgroup, along with a table listing all the applications each user has access to. This includes all the workgroups the user is part of that have access to different applications. "Has Access" will appear for applications the user can access, while a dash will indicate applications the user does not have access to.
You can click on a user's name to open a detailed view showing which workgroups the user is part of and the applications those workgroups have access to.
To invite a user, click the "Invite" button on the right-hand side. This will open a pop-up where you can enter the email address of the user you wish to invite. You can specify more than one email address using a comma-separated list. Note that the email format must match the Allowed Emails specified in the Domain tab.
You can then choose whether the user has access or no access to the different products available to the workgroup. Select the option to "Invite via public Illumina account" if the user you are inviting has a public Illumina account. To invite via collaborative enterprise, you will need to configure the collaboration domains, which can be done under Domain > Collaboration Management. Click "Grant Access" to grant the user access.
To change the access for a user or users, select the checkbox next to their name(s) and click the "Change Access" pencil icon on the right-hand side. The emails of the selected users will appear in the Users dialog. Update the applications the users have access to, and click "Grant Access" to save the changes.
To remove a user or users, select the checkbox next to their name(s) and click the "Remove Access" icon on the right-hand side. A dialog will open where you can confirm that the user should be removed. Click "Remove" to confirm.
Note that workgroup administrators and owners cannot be removed. These users will be skipped from the list of users to remove, and a warning message will be displayed.
To remove a workgroup administrator or workgroup owner, you must first remove their administrator/owner role. Refer to the "Admins" tab above for instructions on how to do so.
In the Pending Invites screen, you can see which users have not yet accepted their invites, as well as the applications and roles they would be granted upon accepting their invitation. From here, you can click "Resend Invitation" to send the invitation again.
Under the Applications tab, you'll see a tab for each application your workgroup has access to. By clicking on an application, you'll see a list of users in the workgroup who have access to it. From this screen, you can invite new users to the workgroup and modify access. See the sections above for information on how to do so.
To manage domain access, click the "Manage Domain Access" button. This will redirect you to the Domain tab. For more information on domain management, refer to the Domain section.
Adapter Read 1, 2 Masking
AdapterRead1,A/T/C/G AdapterRead2,A/T/C/G AdapterBehavior,mask (default trim)
Adapter Read 1, 2 Trimming
AdapterRead1,A/T/C/G AdapterRead2,A/T/C/G AdapterBehavior,trim (default trim)
Adapter Stringency
AdapterStringency,# (default 0.9, 0.5-1.0 allowed)
Barcode Collision Checks
Depending on version, index collision checks are relaxed, strict, or configurable:
3.9.x: Relaxed by default, no option to change. A collision in one index can be rescued by having enough diversity in the combined dual index sequence.
v3.10 and 4.0: Strict by default, no option to change. For a dual index run, requires only 1 index (index1 or index2) to have a collision in order to error out based on a barcode collision. Identical indexes within index1 or index2 (combinatorial dual indexes) still supported.
Barcode Mismatches
BarcodeMismatchIndex1,# (default 1) BarcodeMismatchIndex2,# (default 1)
Combine multiple FASTQ files
NoLaneSplitting,true or false (default false)
Ignore beginning or end of read
OverrideCycles,Y#;N# (default all cycles used) Note: OverrideCycles can be specified in the BCL Convert Settings section to be applied globally or in the BCL Convert Data section to be applied uniquely per-sample.
Index FASTQs
CreateFastqForIndexReads, 0 or 1 (default 0)
Minimum Number of ATCG Bases per Read
MaskShortReads,# (default 22)
Minimum Read Length
MinimumTrimmedReadLength,# (default 35)
Trimming last bases when they match the adapter
MinimumAdapterOverlap,# (default 1, 1-3 allowed) Never trims or masks less than X bases when they overlap with the adapter provided regardless of stringency settings, where X is the MinimumAdapterOverlap provided.
UMI Settings
OverrideCycles,U# TrimUMI,0 or 1 (default 1)
Use a subset of bases for i7/i5 indexes
Use subset of index cycles for demultiplexing by providing shortened sequence in index or index2 column and providing desired length in OverrideCycles setting (default use all index cycles defined in RunInfo.xml)
Adapter Matching Algorithm
FindAdaptersWithIndels,true (default off, false = Sliding Window)
BaseSpace Run Planning provides an interface for planning a sequencing run. Only specific sequencing instruments support Basespace Run Planning. Refer to Sequencer Auto-launch Analyses Compatibility for details.
Navigate to BaseSpace and change to your desired workgroup or personal context. This may require you login to your domain first.
After selecting the appropriate user/workgroup context, navigate to the Runs tab in BaseSpace. Then click the New Run button and select Run Planning.
Use the guided Basespace Run Planning to create a planned run. See the BaseSpace Run Planning documentation for details.
Set the Analysis Location to "BaseSpace/Illumina Connected Analytics" to configure for Cloud analysis.
The configuration input forms vary depending on the instrument type, including the available secondary analysis applications (see Sequencer Autolaunch Analyses Compatibility for details). Subsequent configuration input forms vary depending on the secondary analysis application chosen. The images below demonstrate screens using a NovaSeq X Series instrument with Illumina DRAGEN BCL Convert analysis.
Upon completion of the Run Planning inputs, the sequencing run can be either saved as a planned run in BaseSpace or exported as a Sample Sheet, depending on the instrument type.
Follow the instructions for the sequencer to execute the planned sequencing run for cloud analysis. If the sequencer does not support consuming planned runs from BaseSpace, attach the sample sheet generated by Run Planning to a manual mode sequencing run.
After a sequencing run is started, BaseSpace is used for monitoring the run through completion. Use the BaseSpace Run Summary view to track the run progress.
Refer to Sequencer Run Data in ICA for guidance on viewing the sequencing run data in ICA.
In ICA, the workflow session is launched and a record is present in the Analyses view in the project. The workflow session record can be identified by the User Reference, which will have "ws" preceding the run name.
Check the "status" of the workflow session to monitor progress in ICA. As orchestrated analyses are launched, analysis records will show in the Analyses view and in the workflow session details.
The workflow session details contain an "Orchestrated Analyses" section with the secondary analyses launched by the workflow session listed. Check the "status" of each analysis for progress. In the example below, a BCL Convert pipeline is launched by the workflow session.
The workflow session details also contain links to the BaseSpace Run Summary, which provides further information about the analyses, such as metrics, logs and reports.
The analysis output folder is stored in the ilmn-analyses folder in the project data view. The output folder name can be found in the Analysis Details view in the Output Files section. Filter for the correct output folder by copying the folder name from the Analysis Details view. The image below shows an example output folder from a BCL Convert automated analysis.
Post-sequencing automated analysis will typically include a demultiplex step using the Illumina-provided BCL Convert pipeline. The demultiplexed outputs from this pipeline result in the creation of project sample assets linked to associated demultiplexed data (ie, FASTQs).
With analysis data available in the BaseSpace externally-managed project, the data can be linked to other ICA projects to serve as inputs for running pipelines, etc. Users with access to the BaseSpace externally-managed project can create links from the destination project Samples and Data views. See the Link Project Data and Link Project Sample ICA documentation for details on linking sample data.
The orchestration of auto-launched ICA pipelines is driven by information provided in the sequencing run sample sheet. When leveraging BaseSpace Run Planning to plan a run, the sample sheet is automatically generated. The sample sheet may also be attached to the run manually on the sequencer. The following sections are necessary for powering the autolaunch of secondary analysis after the sequencing run completes.
To retreive the most up-to-date sample sheet template for your instrument, generate an example using BaseSpace Run Planning.
Log into your BaseSpace Sequence Hub account.
Select the Runs tab, and then select the New Run drop-down.
Select Run Planning.
The FileFormatVersion in the [Header] section must be set to 2 to indicate the sample sheet as v2 format.
Sample sheets generated using BaseSpace Run Planning may contain additional optional header fields such as RunName, InstrumentPlatform, and IndexOrientation.
The primary analysis settings consist of the information used for the on-instrument sequencing and demultiplexing of raw sequencing output data. This includes information about reads, indices, primers, etc.
These sections vary greatly depending on the instrument type used and sequencing configuration. The below examples are used for demonstration purposes only.
For demultiplexing the raw sequencing output data, the settings for demultiplexing (ie, BCL Convert) are included. The below examples are used for demonstration purposes only.
A [Cloud_<pipeline_name>_Settings] and [Cloud_<pipeline_name>_Data] setting are provided for each secondary analysis pipeline to be auto-launched after sequencing. The value used for <pipeline_name> can be any value. It's best practice to refer to the analysis type such as "DragenGermline", "BCLConvert", etc. These sections provide input parameters to the pipelines. The contents of these sections, including the columns, may vary depending on the pipeline used for secondary analysis. The below snippet demonstrates using "DragenGermline" as an example.
The [Cloud_Settings] section must be present and include:
Cloud_Workflow value set to ica_workflow_1
BCLConvert_Pipeline set a valid ICA Uniform Resource Name (URN) for demultiplexing
Note: To find secondary analysis URNs for a specific pipeline, click on the pipeline in the
The `<pipeline_name>` used in the `[Cloud_<pipeline_name>_Settings]` and `[Cloud_<pipeline_name>_Data]` must exactly match the value used in the `[Cloud_Settings]` section for the `[Cloud_<pipeline_name>_Pipeline]`.
This section provides a guided example of the end-to-end user flow for a sequencing run for TruSight Oncology 500 on NovaSeq 6000Dx (RUO mode). The workflow includes automated data streaming to cloud and auto-launch of secondary analysis minimizing manual touchpoints. The tutorial covers:
Instrument and BaseSpace configuration
Run Set-up, start and monitoring
Accessing data in ICA and BaseSpace
Follow instructions to configure sequencing run upload to ICA.
On the RUO side of the instrument, select Instrument Settings.
Select RUO SETTINGS tab to configure BaseSpace connection.
Select the box BaseSpace Sequence Hub: (Optional to upload and store run data in cloud server)
Select Run Monitoring and Storage (image below) in the hosting location dropdown menu
Specify the domain name. It is only possible to use a private domain. Contact the IT department to add the correct URLs to the firewall allow list. For further information, please refer to and .
BaseSpace Run Planning provides an interface for planning a sequencing run.
Navigate to and change to your desired workgroup or personal context. This requires you to login to your domain first.
Navigate to the "Runs" tab in BaseSpace. Select the "New Run" button and select "Run Planning".
Use the BaseSpace Run Planning tool to create a planned run.
Set the Analysis Location to "BaseSpace/Illumina Connected Analytics" to configure for Cloud analysis.
The configuration input forms vary depending on the instrument, secondary analysis pipeline, and other selections (see for details). The images below demonstrate screens using a NovaSeq 6000 Dx instrument with DRAGEN TSO500 analysis.
Select "DRAGEN TSO 500 Analysis Software - 2.5.2" or "DRAGEN TruSight Oncology 500 Analysis Software - 2.5.2 (with HRD)" from the Application drop-down menu.
Select TruSight Oncology 500 or TruSight Oncology 500 High Throuput From the Library Prep Kit drop-down menu.
Click Next to navigate to the next step.
Enter the names of the samples in the run. Alternatively, you can download a sample template by clicking "DOWNLOAD TEMPLATE" as indicated in the image below, then use it to import the sample information.
In the Per Sample Configuration section, add Sample Type, Pair ID, and Sample Feature. For HRD sample, specify "HRD" in the Sample Feature field, otherwise leave it empty.
Upon completion, export the sequencing run as a SampleSheet.csv file.
Follow the instructions for the sequencer to execute the planned sequencing run for cloud analysis. Attach the SampleSheet.csv saved from the previous step.
After a sequencing run is started, use BaseSpace to monitor the run through completion. Use the view to track the run progress.
Refer to for guidance on viewing the sequencing run data in ICA.
When secondary analysis finishes, the results can be viewed in BaseSpace in the corresponding project. The result files can be viewed and downloaded from the Files tab. In addition, the files can be downloaded using BaseSpace Sequence Hub CLI (BaseSpace CLI) tool.
"Order Registration" and "Software Registration" terms are used interchangeably.
Thank you for purchasing Illumina Connected Software. To begin using the software, please complete the setup process. This process activates the software and allows you to assign access to specific users. To get started, follow the steps below.
This tutorial will walk through the end-to-end flow for running a NovaSeq 6000 sequencing run with secondary analysis auto-launch in ICA. The secondary analysis will consist of BCL Convert only.
This tutorial is intended to provide guidance for the cloud analysis-related functions. Specific settings and configurations for sequencing will need to be modified for compability.
The main steps in the tutorial consist of:
CombinedIndexCollisionCheck,value Where value corresponds to the number of the lane or lanes to use this behavior; multiple lane values should be semicolon-separated.
4.1.7 and 4.2.x: Relaxed mode by default. CombinedIndexCollisionCheck removed. Mode now configured by using sample sheet option:
IndependentIndexCollisionCheck,value to allow optional strict checking, where value corresponds to the number of the lane or lanes to use this behavior; multiple lane values should be semicolon-separated.
Optional: Enter a description for the run. The run description can contain a maximum of 255 characters.
Select your sequencing system as the instrument platform.
For Secondary Analysis, Select BaseSpace.
Note that if you have an ICA subscription, this option will read BaseSpace / Illumina Connected Analytics.
Optional: Enter the ID for your library tube. The library tube ID is located on the label of your library tube strip.
Select Next.
Select your analysis application. If only one option is available, it will be selected automatically.
Optional: Enter a description for the configuration.
Select an existing library prep kit, or add a new custom library prep kit as follows:
Select Add Custom Library Prep Kit under the Library Prep Kit dropdown.
Enter the name, read types, default read cycles, and compatible index adapter kits for your custom library prep kit.
Select Create New Kit.
Select an existing index adapter kit, or add a new a custom index kits as follows. If you are using more than one library, the libraries must have the same index read lengths.
Select Add Custom Index Adapter Kit under the Index Adapter Kit dropdown.
Select a template type and enter the kit name, adapter sequences, index strategies, and index sequences. Make sure the second index (i5) adapter sequences are in forward orientation.
Select Create New Kit.
If applicable to your application, select a reference genome.
Select Next to configure secondary analysis settings. Detailed instructions for each application and version can be found here.
Select Next to review the Run and Analysis details.
Select Export to save the Sample Sheet to your device.
[Optional] One or more Cloud_<pipeline_code>_Pipeline entries set to a valid ICA URN for secondary analysis.
[Header],
FileFormatVersion,2[Reads],
Read1Cycles,1
Read2Cycles,1
Index1Cycles,1
Index2Cycles,1[Sequencing],
CustomRead1Primer,false
CustomRead2Primer,false
CustomIndex1Primer,false
CustomIndex2Primer,false[BCLConvert_Settings],
SoftwareVersion,0
BarcodeMismatchesIndex1,
BarcodeMismatchesIndex2,
AdapterRead1,
AdapterRead2,
OverrideCycles,[BCLConvert_Settings],
Sample_ID,NS001
Index,T
Index2,A
Lane,1[Cloud_DragenGermline_Settings]
SoftwareVersion,4.1.5
MapAlignOutFormat,bam
[Cloud_DragenGermline_Data]
Sample_ID,ReferenceGenomeDir,VariantCallingMode
<sample_id>,urn:ilmn:ica:region:<region_guid>:data:<data_guid>#<data_path>,None
<sample_id>,urn:ilmn:ica:region:<region_guid>:data:<data_guid>#<data_path>,None[Cloud_Settings]
GeneratedVersion,0.0.0
Cloud_Workflow,ica_workflow_1
BCLConvert_Pipeline,urn:ilmn:ica:pipeline:<pipeline_uuid>#<pipeline_code>
Cloud_DragenGermline_Pipeline,urn:ilmn:ica:pipeline:<pipeline_uuid>#<pipeline_code>








If you already have a user account for Illumina Connected Software, enter those credentials into the login screen. If you do not have an Illumina user account, then select the "Sign up" link to create a new account. You should choose this option if this is the first time you have registered through the Order Registration Portal.
Once you have logged in, you will either be directed straight to the Order setup page (if you created a new account), or you will see your Connected Home page (if you are logging into an existing account).
To access the Orders page from the Connected Home page, select "Orders" from the left navigation menu.
Once you have made it to the Order Registration page, you should see a table displaying all the orders that need to be registered. This includes details such as the Product name, Setup status, Order date, order number, Customer id, Assignees.
If you’ve been given an order code, you can enter it using the "Enter order code" option at the at the top right corner above the table. This will add the order to your account so you can proceed with registration.
For each order awaiting registration, you can assign specific users to set up the software. Click the button “Manage" under Assignees column, a modal window is displayed where you can view the users currently assigned to the order.
To add a new Assignee, you can enter the email address of the person you wish to invite and click "Add". Press the “Confirm” button to send an email to that person containing a link to the Order Registration portal. To remove someone, click the X next to their email address and press "Confirm".
The purchaser of the software cannot be removed from the order, and you cannot remove yourself.
You can review the "Order details" for each order by clicking on the "Order number" link in the table. This information includes the date the order was placed, the order number, the purchaser email, the subscription term, and other key informations about the order. This information is not editable.
To set up the order, select the order you want to setup and click on the "Setup" option next to the "Enter order code" at the top right corner above the table.
You can select multiple orders to set them up at once. The "Setup" option contains software settings. You will need to select a region and domain/workgroup to which the software will be registered. The domain and region cannot be easily changed after the setup is complete.
First, select the region where the software will be registered. This determines the AWS Region in which your software will be installed and run. It is best practice to choose a region that is geographically close to where the software will be used. Most subscriptions are regional and require selecting a region, but consumables (e.g., iCredits) are not, so this step will be skipped for them.
Next, select the domain or workgroup to which the software will be registered.
This step may vary slightly depending on the type of software you're registering:
Software Subscriptions
Examples:
BaseSpace Sequence Hub Professional
ICA Professional, ICA Enterprise, Illumina Connected Insights, Illumina Connected Multiomics
Software Consumables
Examples:
iCredits, Genome Equivalent Samples
For software subscriptions such as BaseSpace Sequence Hub Professional, you will need to select a workgroup during setup. For all other software subscriptions, you will need to select a domain. For software consumables (e.g., iCredits or Sample Analyses), you may choose either a workgroup or a domain to apply them to.
It is important to complete the setup for subscriptions FIRST before registering software consumables. Software consumables require an active subscription for them to be used.
If the account you're logged in with has access to existing domains or workgroups, they will be listed on the page for you to choose from. Selecting an existing domain or workgroup will not affect or modify user access permissions associated with that account.
If you do not have existing domains or workgroups, then select the "Create a new domain" or "Create a new workgroup" button (depending on the type of order you are registering).
Create a New Domain
Note: When creating a new domain, you will be considered the administrator for that domain. The domain administrator can later be changed in the Admin Console.
Upon clicking "Create a new domain", a modal window will be displayed where you can enter the following details for your domain:
Name: Nickname for domain that is unique and easy to identify (ex. “My Company Name’s Domain”). You may only use lowercase alphanumeric, dash (-), max 50 characters and no spaces.
.
The list of allowed emails and email extensions can be updated later in the Admin Console.
Click "Setup" to create the domain. Make sure to select the name of your domain once it's been created.
Once the configuration settings have been selected, press the button at the bottom of the order item to make the software available for use. Upon clicking this button, you will no longer be able to access or modify the order in the Order Registration Portal.
you will receive a welcome notification that includes getting started information about your subscription
At this point, all users in the domain or workgroup should be able to access the software from Connected Home. However, if you have a software which requires adding specific users to the subscription before someone can access the associated application—such as ICM—you can navigate to the Subscriptions page to assign users.
If you have a software which requires every user to be part of a workgroup before they can access the app—such as ICI, Emedgene, and ICM— you can navigate to the Admin Console to create workgroups and add users to the workgroup.
From the Admin Console, you can also assign different domain admins.
See the FAQ for frequently asked questions and answers.
Prepare Sample Sheet
Instrument Run Setup
Run Monitoring
Analysis Monitoring
Review Analysis Output
Follow the BaseSpace Setup instructions to configure BaseSpace to upload run data to ICA.
To plan the sequencing run, navigate to BaseSpace Sequence Hub.
Go to the "Runs" view using top menu navigation
Click the "New Run" button
Click "Run Planning"
Follow the Run Planning input form:
Input the RunName (ie, NovaSeq6000Tutorial)
Set the Instrument Platform to "NovaSeq"
Ensure Secondary Analysis is set to "BaseSpace"
Click "Next"
Complete the next input form:
Set the Application to "Illumina BCL Convert for ICA - 4.1.5"
Set the Library Prep Kit (ie, Illumina DNA Prep)
Set the Index Adapter Kit (ie, Nextera DNA CD Indexes (24 Indexes, 24 Samples))
Click Next
Complete the next input form to configure the settings for the secondary analysis, including settings used for demultiplexing.
Select the Library Prep Kit (ie, Illumina DNA Prep)
Set the Index Reads (ie, 2 Indexes)
Set the Read Type (ie, Paired End)
Set the Read Lengths
Complete the Sample form, including Lane, Sample ID, and index information
(Optional) Add analysis settings
Click Next
Review the planned run on the final screen. Once the planned run is confirmed, click the "Export" button to download the Sample Sheet. The Sample Sheet location will need to be accessible from the instrument (ie, USB Drive, Network Location, etc).
Start the NovaSeq 6000 instrument. Refer to the NovaSeq 6000 documentation for details.
During the Login step, select "Illumina Cloud Options" and "Run Monitoring and Storage" to have the run monitoring and secondary analysis performed in BaseSpace and ICA. Login to the same account used to plan the run and click Next.
Refer to the NovaSeq 6000 documentation for guidance on the Run Setup screen.
Select to "Browse" for the Samplesheet and select the modified sample sheet from the last section. Click Next when complete
Review the run configuration on the final screen. Once confirmed, click to Start Run.
Once the run has been started on the instrument, the run will show in the BaseSpace Runs view after the connection has been established from the instrument to BaseSpace.
Navigate to BaseSpace and select "Runs" in the top menu navigation. Find the run name specified during Run Planning. Click to view the Run Summary view.
Run data will be uploaded to ICA. Navigate to ICA to verify the data is present.
Secondary analysis will start after the sequencing run data has been uploaded. BaseSpace will indicate the analysis starting on the Run Summary view in the "Pending Analyses" list.
Once the analysis has been launched, a workflow session is launched in ICA. This can be found in the BaseSpace externally-managed project where the sequencing run data is uploaded. See Sequencer Run Data in ICA for more information.
Navigate to the "Analyses" view within the project to find the workflow session. As the workflow session runs, the secondary analysis pipelines are launched and show in the "Analyses" view, or in the "Orchestrated Analyses" section of the workflow session details view. See Automated Secondary Analysis in ICA for more information.
The results of the secondary analysis can be viewed in BaseSpace and ICA.
To view the analysis results in BaseSpace:
Navigate to the Run Summary view
Click the Analysis in the Latest Analysis or Prior Analyses sections
Analysis results are shown in the Summary. Navigate to the Files tab to find the output files.
To view the analysis results in ICA:
Navigate to the BaseSpace externally-managed ICA project corresponding to the workgroup/user the run was performed with
Navigate to the Project Analyses
Find the Analysis with the User Reference prefixed with the sequencing run name
Click to view the Analysis details
Copy the User Reference
Navigate to the Project Data
Search the Project Data using the copied User Reference value from step 5
Navigate into the folder resulting from the filter to find the analysis output data
Added Platform Home section with information about Managing API Keys, Subscriptions and more.
Updated Software Setup docs to reflect changes in software registration process.
Added Usage Explorer section with information on how to navigate the new tool and track usage across products.
Updated Sample Sheet documentation.
You can now view sample sheet parameters and definitions by selecting your instrument under the "Instrument Settings" section, choosing the desired pipeline from the right navigation panel, and selecting the appropriate version.
Added a BCL Convert Interactive Sample Sheet, allowing users to click on different parameters to view detailed explanations of each.
Included an and to clarify which i5 sequence orientation to use for different platforms, based on the analysis software.
Added information about various and their compatibility with different instrument platforms.
A new "" section provides additional information on , , and a comprehensive .
Updated documentation
Added information on the /IAM Console.
Updated Sequencer Auto-Launch Analyses Compatibility table
Updated Sequencer Auto-Launch Analyses Compatibility table
Added Secondary Analysis Troubleshooting information for analyses not showing up in ICA
Updated Sequencer Auto-Launch Analyses Compatibility table
Added links to Custom Design Products
Updated user documentation links
Updated Cloud Analysis Auto-Launch page to include instructions for viewing sequencer run data and analysis in BaseSpace
Made changes to page to include updated information for requeuing an analysis in BaseSpace
Updated table
Added page in Reference section
Initial release
Added Software Registration instructions and FAQ
Added links to all documentation sites in the Illumina Connected Software suite
Added instructions
Added for MiSeq
Added comprehensive guide for performing
Created table with guidance on sequencer compatibility with different versions of DRAGEN secondary analysis pipelines and applications with auto-launch capabilities
Added with step-by-step instructions for performing downstream analyses and interpretation in the Cloud





































This tutorial provides a guided example of the end-to-end workflow for a sequencing run on MiSeq with automated launch of secondary and tertiary analysis in Illumina Connected Insights.
The latest v4.1.0 version of MiSeq Control Software (MCS)
Illumina Connected Analytics (ICA) subscription (comes with included BaseSpace subscription)
Illumina Connected Insights subscription
Positive balance of Genome Equivalent Samples and iCredits
At least one workgroup
Personal context is not supported in Connected Insights
From the Main Menu, select System Settings.
Select the Run Settings tab.
Select the BaseSpace tab.
Log in to BaseSpace as the Workgroup Owner and make sure the desired workgroup is selected.
Workgroup Administrators can change the workgroup owner in IAM Console
Click on the workgroup name from the BaseSpace header, select Settings from the expanded menu.
Make sure ICA Run Storage is turned on.
Log in and navigate to Connected Insights, making sure the same workgroup is selected as in the previous steps.
Optional: Navigate to Configuration -> Report Automation and configure report automation with preferred settings. This feature allows for the automatic generation of a draft report based on user settings.
Navigate to Configuration -> Test Definition and create a test definition with preferred settings. Turn on Report Automation if you configured report automation settings in the previous step.
Navigate to Configuration -> Data Upload -> From Illumina Connected Analytics, add the DRAGEN Amplicon v4.2 pipeline, and select the test definition created in the previous step.
Log in to and make sure the same workgroup is selected.
Navigate to the Runs tab, select New Run, and then select Run Planning.
Select Instrument Platform as "MiSeq".
Set the Secondary Analysis to "BaseSpace/Illumina Connected Analytics".
The configuration input forms vary depending on the instrument platform, secondary analysis pipeline, and other selections (see for details). The following images demonstrate screens using a MiSeq instrument with DRAGEN Amplicon analysis.
Select "DRAGEN Amplicon for ICA - 4.2.7" from the Application dropdown menu.
Select the desired library prep kit from the Library Prep Kit dropdown menu.
Select Index Adapter Kit.
Use default values for Index Reads, Read Type, and Read Length or change as needed.
Input Sample IDs and appropriate I7 and I5 indices for each sample. Alternatively, download a sample template and use it to import the sample information.
Under the Analysis Settings section:
Make sure BAM is selected in the Map/Align Output Format field. Note: Connected Insights doesn't support CRAM yet.
Select the DNA BED file matching the Library Prep Kit if DNA samples are included in the run.
Select Somatic from the DNA Caller Variant dropdown menu.
Connected Insights requires sample's disease information to start the analysis. Users can provide disease information in several ways, incorporating disease information into the sample sheet is one of them.
Use a text editor to open the SampleSheet.csv file generated in the previous step.
Find [Cloud_DragenAmplicon_ICA_Data] or [Cloud_DragenAmplicon_Data] section.
Add ,Tumor_Type at the end of the next line after [Cloud_DragenAmplicon_ICA_Data].
From the Home screen, select Sequence.
From the Run Setup Option screen, select Manual.
From the BaseSpace Options screen, select Use BaseSpace™ Sequencing Hub for this run.
Sequencing run results will be uploaded to ICA. Users can monitor the sequencing run status from the instrument or BaseSpace.
Secondary analysis will automatically start upon the sequencing run completion. Users can monitor the secondary analysis status from BaseSpace or ICA.
Cases are automatically created in Connected Insights upon the successful completion of secondary analysis. The tertiary analysis automatically starts if disease information is provided in the sample sheet.
If disease information was not provided in the sample sheet, the case will be created with the "Missing Required Data" status. To start the analysis, follow these steps:
Log in to Connected Insights and make sure the right workgroup is selected.
Open a case with the "Missing Required Data" status.
Click the Edit Case button, then input the tumor type in the Disease field.
Click the Save button. The case will start processing after a few seconds.
For additional options to provide case disease information with the Case Metadata file, access [DATA UPLOAD -> Custom Case Data Upload] in the Connected Insights user guide accessible in the software.
Users can open the case to review the tertiary analysis results after the case finishes processing and the status changes to "Ready for Interpretation". A draft report will be ready for review if the Report Automation feature is turned on.
Use the following steps to requeue secondary analysis in case of its failure:
Delete failed cases in Connected Insights if needed.
Log in to BaseSpace and open the run.
Click the hourglass icon, then select Requeue -> Planned Run.
Choose either the Sample Sheet from the run or upload a new Sample Sheet to requeue the secondary analysis.
New cases will be created in Connected Insights after the secondary analysis completes.
This page includes release notes for Account Management, which apply to all software products. For release notes related to a specific product, please refer to that product's documentation site.
Select the region the instrument should connect to. This setting is used to ensure proper data storage is sent to Illumina Proactive Support and BaseSpace Sequence Hub.
Select Private Domain and enter the private domain of your BaseSpace Sequence Hub account.
Optional: Select or upload a new CNV Panel of Normal file if DNA samples are included in the run and you want to enable the CNV caller.
Optional: Select Yes for DNA Enable SV Calling if DNA samples are included in the run and you want to enable the SV caller.
Select the RNA BED file matching the Library Prep Kit if RNA samples are included in the run.
Select No for RNA Enable Differential Expression.
In Per Sample Configuration, add DNA or RNA and Pair ID. One DNA sample and one RNA sample can be paired and analyzed in Connected Insights in a single case if they share the same Pair ID.
Upon completion, export sequencing run settings as a SampleSheet.csv file.
,. Note: samples sharing the same Pair ID should have the same Tumor Type.Save the file.
Select the Run analysis, collaboration, and storage option to send all run data to BaseSpace Sequence Hub.
Enter your BaseSpace Sequence Hub account credentials.
Select Sign In.
Select the Workgroup.
Select Next.
Set the Read Type, Read Length, and Custom Primers for all of your cycles.
Browse to select the sample sheet CSV file generated and updated in the previous step.
Select Next.
Continue to set up the sequencing run as per the MiSeq System Product Documentation.

















Invisible reCAPTCHA added for sign-up and password reset for better security.
Fixed login reliability issues, including handling special characters and improving error messages.
Full UTF-8 support for names and sign-up field
Software Registration Enhancements
Clearer error messages when exceeding registrant limits.
Performance & Stability
Faster application performance through optimized caching.
Fixed intermittent navigation and order display issues.
Login and Sign Up
Fixed login issues for users with UTF8 characters in passwords. Affected accounts will be automatically repaired upon next login.
Software Registration
Order setup modal now includes a loading indicator during setup operations, improving user experience and clarity.
Restored visibility of order item assignees during multi-item registration workflows.
Improved order registration confirmation message to clearly guide users to check their email for next steps.
Login and Sign Up
Unified Login now features a cleaner interface with automatic SSO redirection and improved keyboard navigation.
Enhanced password flexibility and support for email addresses containing "+".
Software Registration
Multi-order registration is now supported, allowing users to configure and register several orders at once.
Welcome emails are dispatched immediately after registration, speeding up onboarding.
UI improvements include better navigation, clearer messages, and visibility of MFA options when enabled.
Usage and Billing
Fixed a bug where downloading usage reports over 3 months caused indefinite loading. Users are now informed of the 3-month limit.
New Features and Enhancements
Customer Number added to User Registration A new optional "Customer Number" field has been added to the signup form and is now synced with MyIllumina.
Fixed Issues
The email field in the signup form now supports addresses containing the '+' symbol, ensuring compatibility with more email formats.
New Features and Enhancements
Connected Navigation Experience We’ve introduced a unified navigation experience across the platform. The new connected home header and local navigation menus are now available, making it easier to move between key sections based on your access.
SSO and Email Extension Configuration Domain administrators can now configure Single Sign-On (SSO) behavior more precisely. This includes the ability to claim corporate domains, enforce SSO login based on email domains, and manage an exclusion list for users who can still log in with a password.
Improved Order Registration Experience Multiple updates have been made to the software registration page (now called "Orders"), including clearer button labels, improved tooltips, and more intuitive text descriptions to guide users through the setup process.
Streamlined Order Assignment Assigning users to orders is now more efficient. We’ve removed unnecessary email notifications and simplified the assignment flow to reduce clutter and confusion.
Visual and Text Updates We’ve made several UI improvements including updated banner messages, clearer error messages, and more accessible layouts.
Fixed Issues
Login and Authentication
Resolved problems with expired or inactive accounts.
Addressed bugs affecting SSO login.
Added support for + character in usernames.
Fixed case sensitivity issues in email addresses during registration.
User Interface Fixes
Resolved a range of visual bugs and inconsistencies to reduce confusion and improve clarity.
Fixed icon colors, toast messages, and layout glitches introduced in recent updates.
Notification Improvements
Reduced unnecessary email alerts.
Ensured consistent and user-friendly messaging across different user actions.
New Features and Enhancements
You can now select "DRAGEN GB License" as the Usage Type. When this option is selected, the "Measure By" dropdown allows you to view usage by either cost (iCredits) or quantity (gigabases).
Fixed Issues
Fixed an issue where the iCredit balance was not displayed in the Usage Explorer for inactive subscriptions. The balance will now always be shown, regardless of subscription status.
New Features and Enhancements
Defaulted the cost unit to iCredits when no usage data is present.
Fixed Issues
Fixed an issue where the usage context switcher was occasionally not displayed.
Fixed an issue where the Product Dashboard was not displayed after logging in using the new login experience.
Fixed an issue that prevented users from logging in to Illumina cloud software from certain instruments.
New Features and Enhancements
The Usage tab currently available in Admin Console was deprecated and replaced by the new Usage Explorer.
You can now access the Usage Explorer by clicking "Usage Explorer" in the Platform Home dropdown menu.
Removed the low/negative iCredit balance banners in favor of the new "" view
Fixed Issues
Current Domain Owner can no longer remove themself from Domain Administrators
Updates to the Admin Console labels, errors, and tool tips
Fixed issue preventing registration of BaseSpace Professional subscriptions when users were logged with a user other than their public domain user.
New Features and Enhancements
Domain admins can now assign and unassign users to subscriptions on the Platform Home Subscriptions page. This is useful for products that require specific users to be assigned to the subscription in order for someone to access the associated application.
Illumina registration emails now include order codes that users can share. These codes allow any user to self-assign to all pending order items in the Illumina Software Registration Portal.
The Illumina Software Registration Portal link is now accessible from the Product Dashboard top-right dropdown within any domain.
The number of users that can be assigned to pending order items in the Illumina Software Registration Portal has increased from 3 to 20.
You can no longer change domain and workgroup owners in the Software Registration Portal. This action is now available from Admin Console and Platform API.
Software subscriptions for the same product can now be registered to domains with overlapping start and end dates.
A single registration email which includes an order code will now be sent to the users that are automatically assigned to that order (such as the purchaser).
Added pagination for page.
You can no longer change the domain owner (i.e. the primary domain admin) from the Illumina Software Registration Portal. You must go to the Admin Console to update the domain owner. This is now consistent with how workgroup owners are managed.
Workgroup admins can now change workgroup owners in the Admin Console and updates are reflected in the Software Registration Portal.
Fixed Issues
Fixed an edge-case where subscriptions involving apps containing a dot "." would not be processed by Subscription Service properly
Allow special characters in passwords for v1/v2 login
Allow special characters (e.g. "+" and "%") in Admin Console search patterns when included in usernames
Fixed an issue that caused users to be logged out immediately after login
Fixed a rare case where certain users could not be set as workgroup owner/workgroup admin due to email restrictions
Improved workgroup and domain search functionality
Fixed Usage Explorer date range selector when using a non-US region in browser settings
Below you can find an interactive sample sheet with example values filled out for BCL Convert. Click on the dotted underlines for pop-ups with more information on each section.
[Header]
FileFormatVersion
2
RunName
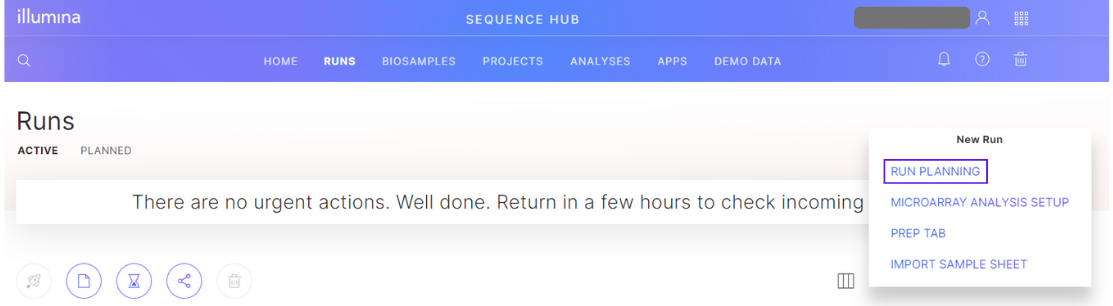
Illumina software usage is measured in two ways:
iCredits: Used for data analysis, computation, storage, and transfer.
Samples: Used for interpreting sequenced samples. Different types exist based on the software and use case, such as:
DummyRun
InstrumentPlatform
NovaSeqXSeries
IndexOrientation
Forward
[Reads]
Read1Cycles
151
Read2Cycles
151
Index1Cycles
10
Index2Cycles
10
[BCLConvert_Settings]
SoftwareVersion
4.1.23
TrimUMI
0
Override Cycles
Y101N50;I10;I10;N50Y101
FastqCompressionFormat
gzip
[BCLConvert_Data]
Lane
Sample_ID
index
index2
1
S01-TOO-12plex-P1-rep1
ATCCACTG
AGGTGCGT
1
S02-TOO-12plex-P1-rep2
GCTTGTCA
GAACATAC
1
S03-TOO-12plex-P1-rep3
GCTTGTCA
GAACATAC
1
S04-TOO-12plex-P1-rep4
GCTTGTCA
GAACATAC
[Cloud_Settings]
GeneratedVersion
1.14.0.202408061842
Cloud_Workflow
ica_workflow_1
BCLConvert_Pipeline
urn:ilmn:ica:pipeline:d5c7e407-d439-48c8-bce5-b7aec225f6a7#BclConvert_v4_1_23_patch1
[Cloud_Data]
Sample ID
ProjectName
LibraryName
LibraryPrepKitName
IndexAdapterKitName
Sample_001
RunName_2024-10-04T23_02_37_72dc905
Sample_001_GCCTTGTCAA_GATCCAATCA
10x3PrimeCellMultiplex
10xIndexNNSetA_3PrimeCellMultiplex
Workgroup owners for BaseSpace Sequence Hub Professional workgroups can now be changed via the Admin Console, consistent with other domain workgroups.
You can no longer set the account unlock time from the Admin Console. It is now fixed at 30 minutes and can only be modified by contacting Illumina Support.
You can no longer add duplicate emails/email extensions to the "allowed emails" for a domain in Admin Console.
Added search box on Admin Console Workgroup Users and Admin pages
Provided more clarity in the error message displayed when an issue occurs while inviting a user to a workgroup.
Domain admins can now view all workgroups a domain user has access to.
You can now see a banner at the top of the Admin Console Usage tab to indicate page retirement on April 30.
Made updates to Admin Console UI and loading messages.
Fixed an issue that allowed user to update the workgroupID for a workgroup they are not a member of
Fixed an issue where domain users without allowed email address are unable to be set as a workgroup owner
Emedgene Array Samples
Illumina Connected Insights Genomic Equivalent Samples
Ensure you purchase the correct samples for your workflow requirements.
Below are the Illumina software products and their software usage pricing details:
Illumina Connected Analytics (ICA) is a cloud-based platform for multi-omics data management and analysis. Charges for storage and compute workloads are in iCredits. For pricing details, visit the ICA Pricing page.
BaseSpace Sequence Hub provides genomic data analysis, storage, and sharing. It supports apps for alignment, variant calling, and data visualization. Costs for storage, compute, and apps are in iCredits. For pricing details, visit BaseSpace Pricing page.
Illumina Connected Multiomics (ICM) is a cloud platform for biologists and bioinformaticians to analyze multiomics data efficiently. It organizes data into studies, applies statistical methods, references biological databases, correlates results across data types, and offers visualization tools for interpretation. Costs for this streamlined data analysis are in iCredits. For pricing details, visit ICM Pricing page.
Illumina Connected Insights (ICI) is a robust software for interpreting and reporting NGS data. It processes DNA and RNA oncology assay data, supporting common tumor variants like SNVs, indels, CNVs, SVs, fusions, and splice variants, as well as biomarkers such as TMB, MSI, and GIS for HRD assessment. By integrating with multiple genomic databases, it provides biological insights and creates tailored summary reports.
There are two ways to access Connected Insights:
Cloud - Costs for cloud analysis are charged in Genomic Samples (GEs) and costs for data storage on ICA are charged in iCredits.
Local - Costs for local analysis via DRAGEN Server are charged in GEs.
For pricing details, visit page.
Emedgene unlocks genomic insights for hereditary disease and streamline your tertiary analysis workflows. The Emedgene platform is divided into two applications:
Analyze - Genomic analysis workbench, where you can accession, interpret, curate and report on your cases, while also efficiently managing the lab workflow
Curate - A repository for all of your organizational curated knowledge
Costs for this analysis are in Genomic Samples(GEs).
All software usage is eligible for charging. This includes any compute, storage, data transfer, or other metered activity associated with your account.
We recommend that you regularly review your usage and understand how costs accrue, both in one-time costs, such as compute events, or ongoing, such as storage. You are responsible for stopping usage and deleting any data/storage you no longer want to pay for.
Billing is usage‑based: if resources continue running or data remains stored on the platform, costs continue to accrue. To avoid charges, please shut down usage and remove data you no longer need.
The Usage Explorer is your central tool for managing and optimizing software consumption at Illumina. It helps you to:
Monitor Usage in Real Time
Track how iCredits or samples are consumed across various products and projects.
Manage Billing Balances
Easily view Pre-Funded and Post-Paid balances at a glance to always stay informed.
Audit and Forecast
Access detailed monthly usage statements.
Generate custom usage reports to analyze trends, forecast future needs, and prevent unexpected costs.
Optimize Behavior
Identify high-usage patterns and adjust workflows to remain within budget.
Software usage requires a subscription and can be billed through either monthly invoicing or pre-funded credits. Software usage differs based on software products and usage types.
We offer two billing options for iCredits.
1. Post-Paid (Monthly Invoicing) Billing:
You pay for usage after the occurs, billing is in monthly arrears.
This is the default billing method.
Invoices are sent to the Bill‑To contact on your account—typically your organization’s Finance or Accounts Payable team, or another designated recipient. Some customers also receive invoices through portals like Ariba or Coupa, depending on their setup. If you need to update the Bill‑To contact or add additional recipients, Support/Customer Care can assist.
If you do not purchase or contract any pre-funded iCredits in advance, your usage will be billed automatically at the end of the month.
If your pre-funded iCredits run out (balance reaches zero), billing will switch to post-paid automatically.
2. Pre-Funded Billing:
You purchase or contract for iCredits in advance and draw down from your balance as you use them. There are two ways to pre-fund.
Pre-paid iCredits
You purchase a fixed amount of iCredits upfront. To view purchasing options for iCredits click here
Usage reduces your balance until it reaches zero.
Before your balance runs out, you’ll need to buy more iCredits or your account will switch to Post-Paid billing automatically.
Open Purchase Order iCredits
You contract for a set quantity of iCredits with an expiration date. To view purchasing options for iCredits
A new Open Purchase Order is required when:
Important: Any iCredit usage beyond your Pre-Funded balance will be billed automatically.
You purchase and register 1,000 worth of pre-paid iCredits before March 1. If you use 800 by mid-month, your remaining balance is 200. Based on your average usage, you’ll likely need to buy additional pre-paid iCredits before the billing period ends (March 31). If your balance reaches zero, any further usage will automatically switch to post-paid billing.
You contract an Open Purchase Order for 10,000 iCredits with an expiration date of June 30. If you use all 10,000 iCredits by May 15, you’ll need to create a new PO to continue using pre-funded billing. If June 30 arrives and the contract expires with unused iCredits, you’ll need to renew or contract a new PO to stay on pre-funded billing. In either case—whether the iCredits are depleted or the contract expires—any additional usage after that point will automatically switch to post-paid billing.
Only available through pre-funded billing.
Pre-paid Samples
You purchase a fixed amount of Samples upfront.
Usage draws down from your balance until it reaches zero.
When your balance runs out, you’ll need to purchase additional Samples to continue usage.
When your account uses iCredits (Post‑Paid / Monthly Invoicing) and Prepaid Samples together, your usage is billed in two parallel streams, depending on the product type.
Usage Explorer
A unified view of your overall software usage.
Balances:
iCredits (Post‑Paid): Shows estimated month‑to‑date charges that will appear on your monthly invoice.
Samples (Prepaid): Shows your remaining Sample balance and the usage drawdown for Sample-based products.
End‑of‑month invoice
iCredits: All usage metered via iCredits for the previous month will be billed in arrears
Samples: There is no invoice for Sample usage.
Tip: Each product clearly indicates whether it consumes iCredits or Samples in its usage details, so you can trace charges and drawdowns back to the source.
Example A — Both consumables in play
You run a pipeline that uses iCredits and a separate workflow that uses Samples.
Mid‑month, your Samples drop from 200 to 0; your iCredit accrued charges continue to grow.
Result:
iCredits: You’ll see the total on your next invoice (post‑paid).
Samples: You will need to buy more Samples.
Example B — Buying more Samples
You purchase Prepaid Samples.
Result:
Your Sample balance updates immediately and future Sample‑metered usage draws down from that balance.
Illumina is flexible in moving between billing options. Below are the rules to how transitioning between billing options works:
New Customers
Automatically assigned to Post-Paid billing.
To switch to Pre-Funded billing, purchase Pre-Paid or contract Open Purchase Order iCredits before using Illumina software.
Any usage before Pre-Funded iCredits are purchased/contracted and registered will be Post-Paid billed.
Existing Customers
Automatically move to Post-Paid billing if Pre-Funded iCredits are depleted.
Can move back to Pre-Funded billing by make another purchase or contract.
Depletion of Pre-Funded iCredits results in automatic switch to Post-Paid billing.
Available only through Pre-Funded billing.
New and existing customers must purchase Pre-Paid Samples in order to use Illumina software products that consume Samples.
Illumina provides a range of automated notifications to help users effectively manage their usage and billing, focusing on low balances, threshold alerts, and invoice triggers.
Usage Explorer Usage Notifications
iCredit Monthly Budget Alerts: Notifications are sent when your configured monthly budget threshold is reached, allowing you to monitor spending and take action before exceeding your planned usage. You can set up or adjust your monthly budget notification at any time in Usage Explorer.
iCredit Pre-Funded Billing Notifications
Low Balance Alerts: Notifications are sent when both Pre-Paid and contracted Open Purchase Order iCredit balances are low.
iCredit Post-Funded Billing Notifications
End of Billing Period Alerts: Invoices are sent at the end of each billing cycle.
Sample Post-Funded Billing Notifications
Sample notifications are planned for release in the upcoming year.
This system ensures users can stay informed and proactive in managing their financial commitments efficiently.
Review active resources (workflows, pipelines, jobs) and stop anything you no longer need.
Delete storage and any temporary data you don’t want to keep.
Download or export anything you need to retain off‑platform.
Billing for any remaining usage (e.g., stored data you did not delete) continues according to your billing option.
If your subscription is expired and you cannot access the product to delete storage or shut down usage, contact Support/Customer Care using the contact information at the bottom of this page. We will help restore access so you can remove data and prevent further charges.
Important: If you do not reach out and your data remains on the platform, usage charges will continue.
Set alerts in Usage Explorer.
Schedule a pre‑expiry cleanup: shut down jobs and delete storage you don’t need.
Use project tags/labels to find and remove old or orphaned resources quickly.
Review Usage Explorer weekly to catch unexpected activity.
Illumina software usage is billed through: iCredits and Samples.
iCredits support post-paid (monthly invoicing) or pre-funded billing.
Samples are pre-funded only.
If pre-funded iCredits are depleted, billing switches to post-paid automatically.
Use Usage Explorer to monitor usage, balances, and forecast spend.
For customer service inquiries, email: [email protected]
Visit our website at: www.illumina.com
Cloud analysis auto-launch refers to the automated launch of secondary analysis by Illumina's cloud platforms after a sequencing run is completed. Coupled with the ability to automatically output sequencing data to a cloud location, the auto-launch functionality minimizes user touchpoints and decreases turnaround time.
Sequencing run management, including run planning, run monitoring, and viewing results, is performed via BaseSpace Sequence Hub (BaseSpace). The post-sequencing secondary analysis (ie, demultiplexing, DRAGEN) is powered by ICA pipelines. The analysis results are stored in an /ilmn-analyses/<sequencer_run_output> subfolder in ICA. See the Sequencer Auto-launch Analyses Compatibility table for available instruments and pipelines.
BaseSpace is used for sequencing run planning and monitoring. See BaseSpace Setup for more information.
ICA is used to store sequencing run data and perform secondary analysis. See for more information.
Run Planning - performed in BaseSpace to configure a sequencing run including choosing instrument type, analysis, and sample settings
Start Sequencing Run - performed on the sequencing instrument to select the planned run and launch the run
Data Upload and Run Monitoring
Further details about each step above is described in the sections below.
BaseSpace settings must be configured to send sequencing run data to ICA prior to sequencing. For workgroups, this must be configured by the workgroup administrator. Workgroup users have read-access to the toggle switch setting, but cannot modify it.
Navigate to the BaseSpace Settings view. In the "Settings" section, set the "ICA Run Storage" setting to ON.
This setting is not necessary for NovaSeq X Series instrument type
The visibility of the "ICA Run Storage" configuration setting is controlled by Illumina support. Contact Illumina Support to request the setting be made available for your domain.
Sequencing run planning in BaseSpace Sequence Hub is the first step in preparing a sequencing run with automated post-sequencing analysis.
A sequencing run can be started from the instrument using either:
Planned sequencing run setup in Basespace Run Planning (recommended). See for a guided example.
Sample Sheet imported during the on-instrument Run Setup. See for more information.
Refer to for guidance on sequencer compatibility with different versions of analyses applications.
Monitoring the status of an ongoing sequencing run is done through BaseSpace. See the BaseSpace help documentation for more information.
Find the sequencing run in BaseSpace Sequence Hub by logging in, selecting the Runs tab, and selecting the appropriate sequencing run from the list.
When the sequencing run completes uploading, the run status in BaseSpace Sequence Hub will move to Pending Analysis. Analysis will automatically launch based on the information in the Sample Sheet.
If there is an issue with the Sample Sheet, the status will move to Needs Attention. Use the Requeue feature to correct any issues and re-launch analysis. See
Sequencer run data is stored in ICA in an created by BaseSpace. Operations in BaseSpace, including planning and monitoring instrument runs, are performed in a given user or workgroup context. To preserve permission boundaries between applications, an externally-managed project is created for each user/workgroup context per region. The ICA project will be named using the convention BSSH {context name}, where {context-name} is the user or workgroup name operating in BaseSpace. For workgroups, the project will be created on behalf of the workgroup owner. The project Team includes an entry for the workgroup to grant all workgroup users read-only access to the project. While Team members may be assigned the Contributor role in the externally-managed project, they will not have write access (ie, create/delete data, run pipelines) in the project.
There may be situations when an analysis needs to be launched manually in ICA, such as if the auto-launched analysis failed. Analyses can be launched manually in ICA with a few extra steps to prepare the input data.
For example, if the demultiplexing analysis fails due to a sample sheet mistake or a system error, the analysis can be launched manually in ICA by following these instructions:
There may be situations when an analysis needs to be launched manually with the ICA API if the auto-launched analysis failed. Use the API to create an analysis and set the input type to basespace (for example /api/projects/{projectId}/analysis:nextflow with analysisInput > inputs > externalData > type:basespace) This works for both files and folders.
Secondary analysis failures may happen for a variety of reasons. When an error occurs, the Sequencing Run in BaseSpace will indicate a failure with the analysis. Depending on the specific failure, the workflow session and/or the orchestrated analysis in ICA will indicate a "Failed" status with an Error output in the details view.
Staring with BaseSpace 7.29, there is now improved error reporting through BaseSpace. A link in the logs section of the Analysis Summary tab opens a modal displaying a list of failed steps and relevant log file contents.
If no BaseSpace-managed runs appear in ICA, verify in that the workgroup used in BaseSpace has access to ICA.
Ensure that the option Has Access is selected in as the option Has Access+Archive has been decommissioned
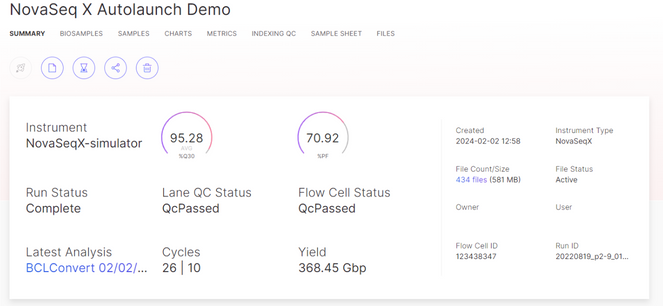
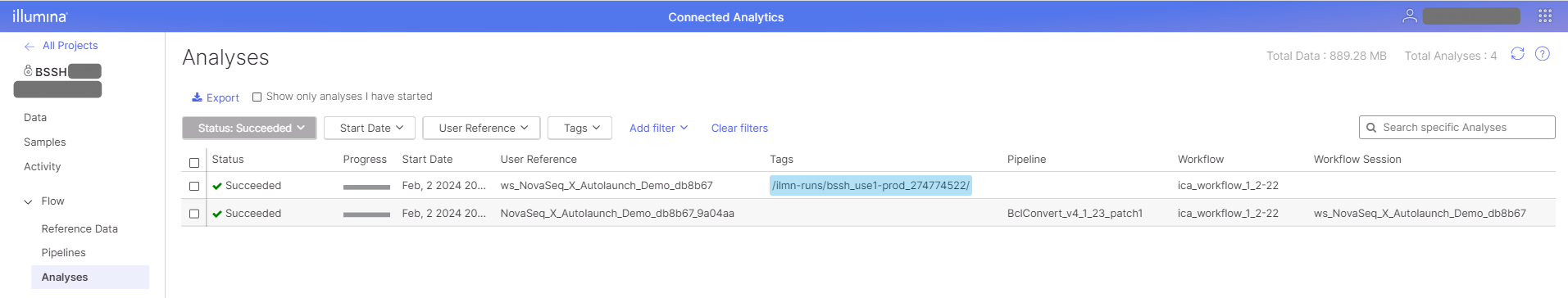



The allocated quantity is used up, or
The contract expires, whichever happens first.
Early renewal ensures uninterrupted pre-funded billing.
The Sample purchase appears as a prepaid transaction (not as post‑paid usage).
Data Upload - the sequencing instrument uploads the sequencing run output data to ICA
Run Monitoring - sequencing run monitoring is available in BaseSpace
Auto-launch Analysis - the secondary analysis is automatically launched in ICA upon sequencing run completion
Auto-launch Analysis Monitoring - the secondary analysis status is monitored in BaseSpace
Edit & Requeue SampleSheet to trigger #4 (optional) - if an error occurs during secondary analysis, the analysis may be requeued (re-launched) from BaseSpace
When the analysis launches, the run status will move to Analyzing. The associated analysis can be found on the Run Details tab under Latest Analysis, or in the Analysis list.
Click the analysis link to go to the Analysis Details page. Information about the analysis progress will be displayed in the Logs section.
If the analysis aborts with an error, logs and any files that had been produced prior to the error will available in the Files tab.
When the analysis completes, any aggregate and/or sample reports can be viewed in the Reports tab. Logs and other output files can be viewed in the Files tab.
The project contains the following tags:
Technical Tags: bssh.project.user:{user/workgroup ID}
For workgroups, users who are members of the workgroup will have access to the data in the externally-managed project.
All sequencing run data is stored in the ilmn-runs folder. Within that folder, the data for each sequencing run is stored in a sub-folder named based on the run name used to create the run during run planning (or as specified in the RunName row of the [Header] section in the sample sheet) appended with a unique identifier.
The screenshot below provides an example of a sequencer run output folder uploaded to ICA.
Sequencer run data uploaded directly to ICA via CLI or Service Connector to a user-managed project will not be available in BaseSpace.
Sequencer run data stored outside of ICA or BaseSpace may be uploaded using the BaseSpace CLI (command-line interface) to make it available in both BaseSpace and ICA.
Use the BaseSpace Uploading a run instructions to upload a sequencer run output folder.
When the sequencing run data upload has completed, the post-sequencing secondary analysis described in the sample sheet will be automatically started.
In ICA, secondary analyses launched after a sequencing run completes is orchestrated by a parent process called a Workflow Session. The workflow session serves as a parent process orchestrating the secondary analysis, including launching secondary analysis pipelines and creating sample entities in ICA.
Upon completion of the sequencing run data upload, a workflow session record is created and visible in the ICA "Analyses" view. The workflow session record can be identified by the User Reference, which will have "ws" preceding the run name.
The workflow session details view includes a section Orchestrated Analyses to indicate analyses launched by the workflow session as part of the automated secondary analysis. These orchestrated analyses are driven by the information in the sample sheet, including the pipeline to launch, reference data, and sample-specific settings.
Currently, only a limited set of Illumina-provided pipelines are compatible with workflow session automation.
For users intending to use BaseSpace for FASTQ generation and DRAGEN Secondary Analysis, the data will be associated with several BaseSpace Projects.
1. SampleSheet ProjectName
When using ProjectName values in Run Planning, the FASTQ files will be written to those locations.
In the SampleSheet.csv, this will be reflected in the [Cloud_Data] section.
Once BCL Convert (and any associated downstream analysis) is completed, the FASTQ files will be associated with the ProjectName specified on BaseSpace. The project column is not required to be specified, and if left blank, the FASTQ Files will be written to a BaseSpace project following the nomenclature:
2. bssh_[region]_[run ID]
For example:
bssh_use1-prod_277283070
More information about regions can be found in the BaseSpace Help center article . The run ID corresponds to the numerical value observed in the web address bar when viewing the run.
If any DRAGEN Secondary analysis is performed, this project will also contain the DRAGEN output.
Follow instructions to Link Project Data to link the sequencing run output data to the user-managed project data
The "ilmn-runs" folder contains all sequencer run data managed by BaseSpace and should not be linked to a user-managed project. Only link individual sequencer run output folders within the ilmn-runs folder. See Sequencer Run Data for details on how to find the correct folder to link.
Link the Bundle containing the ICA pipeline to the user-managed project (ie, "DRAGEN Analysis 4.1.7 - Sequencer Integration Only") (See Link Bundles)
Fix the sample sheet by downloading the original sample sheet from the sequencing run output folder, modify, and re-upload the file to the user-managed project
Launch the pipeline (ie, BclConvert_v4_1_7) with the corrected sample sheet as input
The secondary analysis for a sequencing run may need to be re-executed for a variety of reasons including:
Fix a Sample Sheet error
Intermittent analysis failure
The operation to manually relaunch the secondary analysis is referred to as "requeue". Requeues are performed from BaseSpace Sequence Hub.
This instruction below can be used to requeue auto-launch analysis for NovaSeq X/X Plus, NovaSeq 6000, NextSeq 500/550, NextSeq 1000/2000.
Requeue > Sample Sheet will load the original sample sheet and can be modified directly in the browser. This is for advanced users and less validation of the sample sheet will be performed than if using Requeue > Planned Run.
To requeue autolaunch analysis in the BaseSpace UI, follow the steps:
Click on your run in the "Runs" tab. From the Run Summary Page, select Status > Requeue > Planned Run.\
Upload your new, corrected Sample Sheet. This should be the v2 sample sheet that is used for autolaunch.





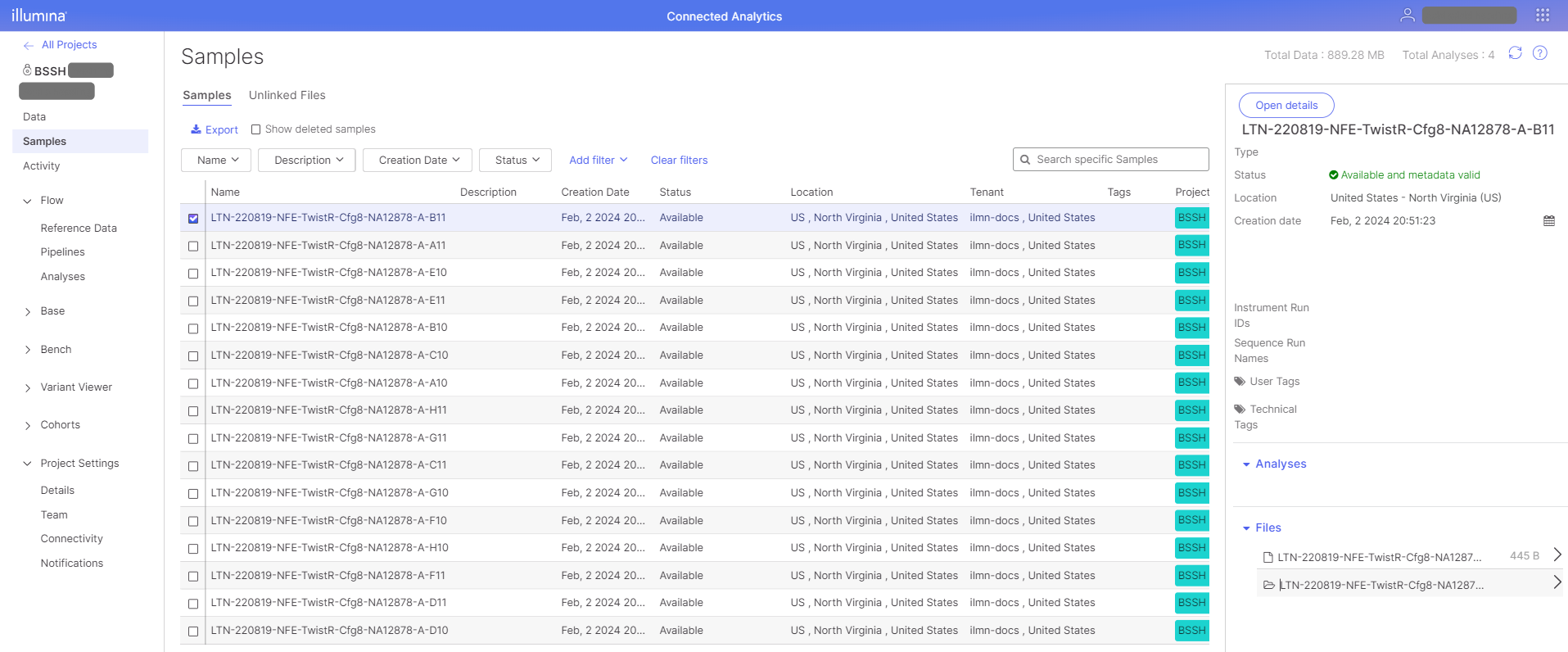
{
"externalData": [
{
"url": "https://api.servername.illumina.com/v2/files/your_files",
"type": "basespace",
"mountPath": "/location_where_input_file_is_saved_on_machine_running_pipeline",
"basespaceDetails": {
"workgroupId": "wid:123" (Optional workgroup id to filter on)
"extensions": "vcf" (Optional BSSH API query param to filter based on file extensions)
"pathPrefix": "/path/to/files" (Optional BSSH query string param to filter files/folder)
}
}
]
}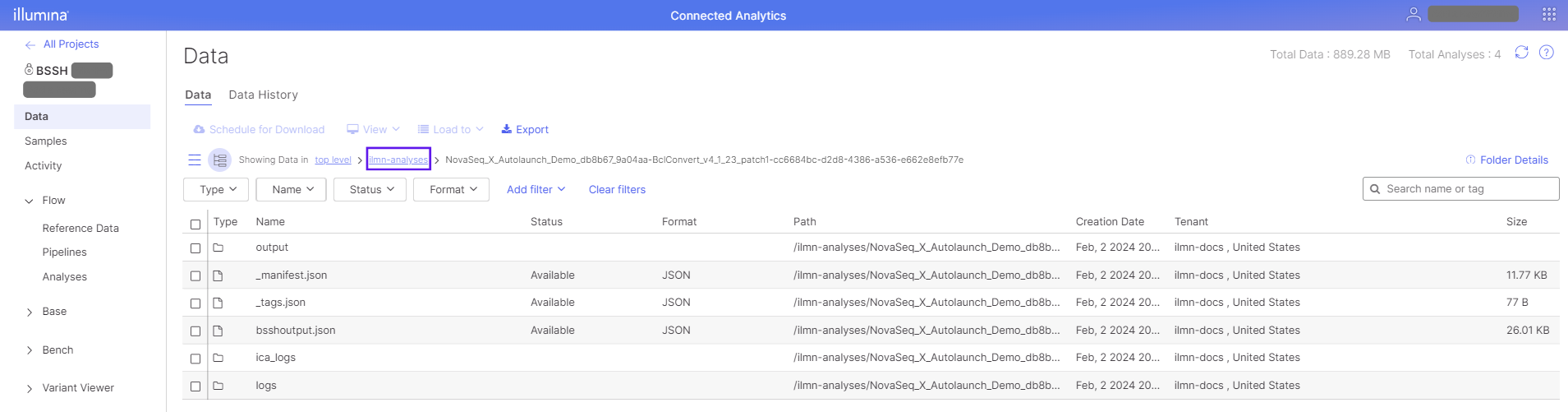








The Domain section displays domain usage and allows you to manage permissions at the domain level. Please refer to the sections below for more information on each tab.
The Usage Report section allows you to generate various usage reports.
You can choose from the following report options:
You can select the days you want to include in the Usage report. Enter the email addresses of the users you wish to send the report to. A notification will appear, letting you know that the report will be sent to your email once it's complete.
Note: Usage reports can only be generated for the last 90 days. For any information older than 90 days, please contact Illumina Support.
Note: If you have your Authentication Type configured to "SAML" Single Sign-On in the Authentication tab, this section will not be visible, and your configured SAML provider's password settings will be used.
In the Password Management section, you can configure different requirements for the passwords users generate when accessing the domain.
These settings are designed to ensure passwords are as strong as possible.
Choose a minimum password length between 8-10 characters.
By default, the option to require one or more special characters is selected.
By default, the option to require at least one digit is selected.
These settings control locking the account after too many unsuccessful login attempts.
Max unsuccessful tries: Choose the number of failed login attempts that will trigger an account lock.
Reset time for lock-up account: Choose the amount of time a user's account will remain locked after the specified number of incorrect password attempts before they can try logging in again.
This setting ensures that a user cannot reuse the same password too many times. Users will be required to set a new password within a set period.
Select the number of days to look back when checking for previously used passwords. For example, if you set this to 30 days, passwords used more than 30 days ago can be reused when a user is required to set a new password.
Choose the number of last-used passwords that should not be allowed as a new password.
The password policy is a text message displayed as an alert during the password selection process on the registration page.
In the Session Management section, you can configure settings for a user's session and inactivity timeout.
These settings allow you to configure the idle session timeout and JWT expiration time.
User's idle session timeout in minutes: Set the number of minutes a session can be idle before it times out. An idle session is one where the user is not actively interacting with the session. For example, if a user is working in a different tab and does nothing in their session, this would be considered an idle session. Accepted values are 5-60 minutes. To disable session timeout, set this value to -1.
JWT expiration time in minutes: This setting determines the duration before the JWT token expires. The JWT (JSON Web Token) is used for securely transmitting information between parties, often for authentication and authorization purposes. It contains claims (such as user information) and is used to verify the user's identity for access to resources. Accepted values are 120 to 10,080 minutes (2 hours to 7 days).
These settings allow you to configure the expiration and active limit for the API key.
API Key expiration time in days: Set the number of days before the API key expires. To disable API key expiration, set this value to -1.
Active API Key limit: Set the maximum number of active API keys that can be used at the same time for a user. This helps manage the number of concurrent API sessions a user can have, ensuring that resources are not overwhelmed by too many simultaneous requests.
In the User Management section, you can manage users, administrators and service accounts.
In the Users tab, you can view a list of all domain users. You can search for a specific user using the search bar at the top.
Click "Manage" next to the user to view more details below the overview table, including information about the user's state and API keys.
In the User State overview, you can view details about the user and expire the account by selecting the "Expire User" checkbox.
In the User API Keys overview, you can view details of the user's API keys, including the name, status, creation date, expiry date, and the last time the key was used. To delete an API key, click the "Delete" button. Click the three dots next to the "Delete" button to view the roles associated with the API key. Make sure to click "Save" to apply any changes.
In the Allowed Emails tab, you can specify which email addresses are allowed to join the domain. To add an email, type the emails or email suffixes, separated by commas, and click the "+" icon. Any allowed emails will appear at the bottom. To remove an email, click the red "X" next to it.
Allowed Email Suffixes: This option allows users to join the domain if their email contains a specified allowed suffix. Do not include the "@" symbol. It is not recommended to allow common email suffixes, such as gmail.com.
Allowed Emails: This option allows users to join the domain if their email exactly matches one of the allowed emails.
The Administrators tab is used to manage the domain's administrators and owners. You can view an overview of the names and emails of users who are currently administrators of the domain.
To remove an administrator, click the "remove" link on the right.
To add a domain owner, click the "Change Settings" link next to "Domain Owner". The domain owner is the primary administrator and main point of contact for users within the domain. They receive monthly emails about iCredit balances and are automatically assigned to any new orders placed for the domain. Assigning a domain owner will automatically make them a domain administrator and user. Once the domain owner field is filled, the domain owner cannot be removed, only replaced. You can update the domain owner at any time by entering a different email address. Note that the previous domain owner will remain a domain administrator.
To add or promote a user to an administrator role for the domain, click "Configure an Administrator," then enter the email address of the user and click "Check." You can then fill out the form and click "Save" to assign the user as an administrator.
In the Domain Invitation tab, you can add one or more user emails to invite them to the domain. Be sure to separate the emails with a comma. Click "Invite" to send the invitations. An overview table will display each user's username, email, and the status of whether they have accepted the invitation. You can revoke their invitation by clicking the "Revoke" button.
In the Service Accounts tab, you can view the service accounts that have access to your domain. Service accounts are special accounts used by applications or services to interact with your domain without requiring a user to log in.
Click the checkbox to block service accounts if you want to disable existing service accounts and prevent the creation of new ones.
You can click "Manage" next to a service account to view more details below the overview table, including information about the account state and API keys. In the Account State overview, you can view details about the service account, such as its activation and expiration dates. Click the checkbox to expire the service account.
In the API Keys overview, you can view details of the account's API keys, including the name, status, creation date, expiry date, and the last time the key was used. To delete an API key, click the "Delete" button. Click the three dots next to the "Delete" button to view the roles associated with the API key. Make sure to click "Save" to apply any changes.
In the Access Management section, you can set IP addresses or CIDR blocks to restrict access to the application to only the specified addresses.
Using IP range-based authentication, you can control access by IP address, allowing or blocking access to specific addresses or ranges of addresses. Examples include 192.10.10.1, 192.255.10.*, or 192.10.10.0/32. Be sure to separate multiple addresses with commas.
Select a preferred method of access management:
Create an allow-list for IP addresses/CIDR to allow access: The IP addresses/CIDR block ranges you enter will be allowed access to the domain.
Create a block-list of IP addresses/CIDR to block access: The IP addresses/CIDR ranges you enter will be blocked from accessing the domain.
In the Authentication section, you can configure the authentication type.
To enable logging into the platform using your organization's identity provider (IdP), a SAML configuration may be provided in the Authentication Configuration for the Illumina domain.
To configure SSO, follow these steps:
Verify your organization's DNS domain in Illumina Connected Software (ICS).
Create a SAML 2.0 application in your IdP.
Configure ICS with your IdP metadata and attribute mappings.
Must be configured with a domain administrator account for your Illumina domain
Access to your IdP to configure the SP application
Your IdP configurations
Step 1: Create DNS Domain record
Go to the for your Illumina domain, and navigate to the Domain tab
Navigate to the DNS Domain Management menu.
Enter your domain (e.g. company.com) and click Add
Step 2: Verify DNS Domain
To confirm domain ownership, add a TXT record to your Domain Name System (DNS) host using the TXT Record Value. DNS propagation can take up to 72 hours. Illumina Connected Software automatically checks for the record during this time.
To add your TXT record to AWS, see .
Wait up to 72 hours for TXT record verification.
After the record is live, go to DNS Domain management in the Admin Console and select Verify.
Step 1: Create SSO connection in IdP
The Illumina Connected Software service provider (SP) application uses the following configuration:
Entity ID: https://login.illumina.com/saml-service/saml/metadata
ACS (Assertion Consumer Service) URL: https://login.illumina.com/saml-service/saml/SSO
Binding: HTTP-POST
Sign in to your Okta account and open the Admin portal.
Select Administration and then Create App Integration.
Select SAML 2.0, then Next.
Step 2: Connect Illumina Connected Software to your IdP
Complete the integration by pasting your IdP values into Illumina Connected Software
Go to the for your Illumina domain, and navigate to the Domain tab.
Navigate to the Authentication menu and enable the SAML Authentication Type.
In Okta, select your app and go to View SAML setup instructions.
Copy the Identity Provider Single Sign-in URL.
Copy and paste into a text editor the IDP Metadata. Save the file.
Allow 15 minutes for the Illumina Service Provider to update with the provided information. To confirm SAML configuration changes, attempt to login using a qualified e-mail (e.g. @company.com).
In the Collaboration Management section, you can configure the collaboration domain namespaces.
This allows you to invite users to a workgroup via Collaborative Enterprise. Enter the domain namespace and click the + button to add it.
In the Role Management section, domain admins can create and manage custom roles with unique permission settings that provide access control within workgroup.
Users should be cautious while applying custom roles as incorrect setup might lead to restricted access and unexpected issues.
You can search by application name or role name at the top. In the overview table, you'll see a list of applications along with their associated roles. You can click on each role to view detailed information via a hyperlink.
To create a new role, click the "Create role" button. Select an application from those available for custom roles, enter role name and a role description (optional), and select at least one permission for setup. You can edit the role after saved. Selecting application is not available for editing.
Note: If you have your Authentication Type configured to "SAML" Single Sign-On in the Authentication tab, this section will not be visible, and your configured SAML provider's MFA configuration will be used.
In the Multi-Factor Authentication section, you can enable Multi-Factor Authentication (MFA) for the domain by selecting the checkbox.
You can specify the number of allowed days to skip MFA setup. This means users will have a grace period (specified number of days) to complete their MFA setup before it becomes mandatory.
You can also set the maximum number of unsuccessful attempts from the dropdown menu. This controls how many failed MFA attempts are allowed before further actions, such as account lockout or additional security measures, are triggered.
This tutorial provides a guided example of the end-to-end workflow for a sequencing run on NextSeq 550 or NextSeq 500Dx (RUO Mode) with automated launch of secondary and tertiary analysis in Illumina Connected Insights.
The latest v4.2.0 version of NextSeq 550 Control Software (NCS)
Illumina Connected Analytics (ICA) subscription (comes with included BaseSpace subscription)
Illumina Connected Insights subscription
Positive balance of Genome Equivalent Samples and iCredits
At least one workgroup
Personal context is not supported in Connected Insights
From the NextSeq Control Software home page, navigate to Manage Instrument.
Select System Configuration.
Select BaseSpace Sequence Hub Configuration.
Log in to BaseSpace as the Workgroup Owner and make sure the desired workgroup is selected.
Workgroup Administrators can change the workgroup owner in IAM Console
Click on the workgroup name from the BaseSpace header, select Settings from the expanded menu.
Make sure ICA Run Storage is turned on.
Log in and navigate to Connected Insights, making sure the same workgroup is selected as in the previous steps.
Optional: Navigate to Configuration -> Report Automation and configure report automation with preferred settings. This feature allows for the automatic generation of a draft report based on user settings.
Navigate to Configuration -> Test Definition and create a test definition with GRCh37 human reference genome. Turn on Report Automation if you configured report automation settings in the previous step.
Navigate to Configuration -> Data Upload -> From Illumina Connected Analytics, add the DRAGEN TruSight Oncology 500 Analysis Software v2.5.2 (with HRD) pipeline, and select the test definition created in the previous step.
Log in to and make sure the same workgroup is selected.
Navigate to the Runs tab, select New Run, and then select Run Planning.
Select Instrument Platform as "NextSeq 500/550".
Set the Secondary Analysis to "BaseSpace/Illumina Connected Analytics".
The configuration input forms vary depending on the instrument platform, secondary analysis pipeline, and other selections (see for details). The following images demonstrate screens using a NextSeq 550 instrument with DRAGEN TruSight Oncology 500 Analysis Software v2.5.2 (with HRD) analysis.
Select "DRAGEN TruSight Oncology 500 Analysis Software - 2.5.2 (with HRD)" from the Application dropdown menu.
Select "TruSight Oncology 500" from the Library Prep Kit dropdown menu.
Select "TruSight Oncology 500 (NovaSeq6000Dx with SP Flow Cell, NovaSeq6000, NextSeq)" from the Index Adapter Kit dropdown menu.
Use default values for Index Reads, Read Type, and Read Length or change as needed.
Input Sample IDs and appropriate Index IDs for each sample. Alternatively, download a sample template and use it to import the sample information.
In Per Sample Configuration, add Sample Type, Pair ID and Sample Feature. One DNA sample and one RNA sample can be paired and analyzed in Connected Insights in a single case if they share the same Pair ID.
Connected Insights requires sample's disease information to start the analysis. Users can provide disease information in several ways, incorporating disease information into the sample sheet is one of them.
Use a text editor to open the SampleSheet.csv file generated in the previous step.
Find [Cloud_TSO500S_Data] section.
Add ,Tumor_Type at the end of the next line after [Cloud_TSO500S_Data].
Add code for each sample following a ,
From the NextSeq Control Software Home screen, select Experiment.
On the Select Assay screen, select Sequence.
On the Run Setup screen, select Manual as run mode.
Sequencing run results will be uploaded to ICA. Users can monitor the sequencing run status from the instrument or BaseSpace.
Secondary analysis will automatically start upon the sequencing run completion. Users can monitor the secondary analysis status from BaseSpace or ICA.
Cases are automatically created in Connected Insights upon the successful completion of secondary analysis. The tertiary analysis automatically starts if disease information is provided in the sample sheet.
If disease information was not provided in the sample sheet, the case will be created with the "Missing Required Data" status. To start the analysis, follow these steps:
Log in to Connected Insights and make sure the right workgroup is selected.
Open a case with the "Missing Required Data" status.
Click the Edit Case button, then input the tumor type in the Disease field.
Click the Save button. The case will start processing after a few seconds.
For additional options to provide case disease information with the Case Metadata file, access [DATA UPLOAD -> Custom Case Data Upload] in the Connected Insights user guide accessible in the software.
Users can open the case to review the tertiary analysis results after the case finishes processing and the status changes to "Ready for Interpretation". A draft report will be ready for review if the Report Automation feature is turned on.
Use the following steps to requeue secondary analysis in case of its failure:
Delete failed cases in Connected Insights if needed.
Log in to BaseSpace and open the run.
Click the hourglass icon, then select Requeue -> Planned Run.
Choose either the Sample Sheet from the run or upload a new Sample Sheet to requeue the secondary analysis.
New cases will be created in Connected Insights after the secondary analysis completes.
Click Next to continue.
Upon completion, export sequencing run settings as a SampleSheet.csv file.
Save the file.
Select Next.
Enter a run name of your preference.
Optional: Enter a library ID of your preference.
From the Recipe drop-down list, select a recipe. Only compatible recipes are listed.
Select Paired-End as read type.
Enter the number of cycles for each read in the sequencing run using the same value as the sample sheet generated from previous step.
Set the output folder location for the current run. Select Browse to navigate to a network location.
Select Browse to navigate to a sample sheet.
Select Purge consumables for this run. The setting purges consumables automatically after the current run.
Select Next.
Select Next to start the sequencing.










SAML Attributes for EmailId, firstName, LastName
To add your TXT record to Google Cloud DNS, see Verifying your domain with a TXT record.
Wait up to 72 hours for TXT record verification.
After the record is live, go to DNS Domain management in the Admin Console and select Verify.
To add your TXT record to GoDaddy, see Add a TXT record.
Wait up to 72 hours for TXT record verification.
After the record is live, go to DNS Domain management in the Admin Console and select Verify.
Sign in to your domain host.
Add a TXT record to your DNS settings and save the record.
Wait up to 72 hours for TXT record verification.
After the record is live, go to DNS Domain management in the Admin Console and select Verify.
NameID Format: urn:oasis:names:tc:SAML:1.1:nameid-format:emailAddress
Name your app "Illumina Connected Software".
Optional. Upload a logo.
Paste the service provider configuration values from above:
ACS URL -> Single Sign On URL
Entity ID -> Audience URI (SP Entity ID)
Configure the following settings:
Name ID format: EmailAddress
Application username: Email
Update application username on: Create and update
Under Attribute Statements enter the following Name->Value attributes for the email address, first name, and last name. Make sure the Name format is set to “URI Reference.”
email -> user.email
first -> user.firstName
last -> user.lastName
Select Next.
Select the This is an internal app that we have created checkbox.
Select Finish.
Sign in to Microsoft Entra (formerly Azure AD).
Select Default Directory > Add > Enterprise Application.
Choose Create your own application, name it "Illumina Connected Software", and choose Non-gallery.
After creating your app, go to Single Sign-On and select SAML.
Select Edit on the Basic SAML configuration section.
Edit Basic SAML configuration and paste values from above:
Entity ID -> Identifier
ACS URL -> Reply URL
Save the configuration.
From the SAML Signing Certificate section, download the Federation Metadata XML.
Return to the Illumina Connected Software Admin Console.
Paste the Sign-in URL in the IdP URL field
Upload the IDP Metadata file from Step 3 to the "Select SAML Configuration File" file uploader.
Add the SAML Attribute Mappings
EmailId -> email
Last name -> last
First name -> first
Review and select Save.
In Entra ID, copy the Login URL from the Configuration URLs
Return to the Illumina Connected Software Admin Console.
Paste the Login URL in the IdP URL field.
Upload the Federation Metadata XML file to the "Select SAML Configuration File" file uploader.
Add the SAML Attribute mappings:
EmailId -> http://schemas.xmlsoap.org/ws/2005/05/identity/claims/userPrincipalName
Last name -> http://schemas.xmlsoap.org/ws/2005/05/identity/claims/surname
Review and select Save.
General Usage Report
View this domain's users, total user sessions, last login details, access counts, registration date, and usernames
Login Report
View account activity for this domain, including client IP addresses, the applications accessed, event types, and user emails
Workgroup Report
View this domain's workgroup activities, including event data for actions performed by each user
Default
The default setting allows the Illumina Authentication System to manage user credentials.
SAML
Users are redirected to your Identity Provider (IdP) to authenticate via SAML 2.0. (see Single Sign-on below).





















This page provides guidance on sequencer compatibility with different versions of DRAGEN secondary analysis pipelines and applications with auto-launch capabilities. This includes:
Pipelines with cloud analysis auto-launch. This type of workflow is available for BaseSpace and ICA users and includes automatic launch of secondary analysis in the cloud after sequencing completion.
Applications installed onboard of sequencers and on DRAGEN servers paired with sequencers. These type of applications allow you to configure settings for the run and analysis upfront and launch secondary analysis automatically after sequencing completes.
Tertiary analysis compatibility. Click on underlined versions in the table to view details on pipelines’ compatibility with Emedgene and Illumina Connected Insights.
This table does not cover all DRAGEN pipelines and applications. If a pipeline is not included in the table, it means that the auto-launch may not be available for it at all or not available for a specific instrument/pipeline combination. In this case, the pipeline could be used with the instrument if launched manually in BaseSpace, ICA or on the DRAGEN server. Non-DRAGEN pipelines are not included. Please check and other resources on for additional onboard applications.
Cloud
Onboard
Cloud
Paired server
Cloud
Cloud
Cloud
Onboard
Cloud
Cloud
Cloud
BCL Convert
v4.2.7 v3.10.4 v3.8.4 v3.7.4 v3.5.8 v3.5.6
v4.2.9
v4.2.7 v3.10.12 v3.10.11 v3.9.3 v3.8.4 v3.7.4 v3.5.8 v3.5.6
v4.3.13
v4.1.23 v4.1.7
v4.3.13
v4.1.23 v4.1.7 v4.1.5
v3.8.4
v3.9.5
v3.8.4
v1.0.31
v1.0.31
DRAGEN Germline
v4.2.9
v4.2.7
v4.2.7 v3.10.12
v4.3.13
v4.1.23
v4.3.13
v4.1.23
v3.9.5
DRAGEN Enrichment
v4.2.7
v4.2.7
v4.3.13
v4.3.13
DRAGEN Amplicon
v4.3.13
v4.2.7
v4.3.13
v4.3.13
v4.3.13
v4.3.13
DRAGEN TruSight Oncology 500
v2.6.0
v2.6.0
v2.6.0
v2.5.2
v2.6.0
v2.6.0
v2.5.2
v2.6.0
v2.5.2
DRAGEN TruSight Oncology 500 ctDNA
v2.6.1
v2.6.0
v2.6.1
v2.6.0
v2.6.1
v2.6.1
v2.6.0
DRAGEN RNA
v4.2.9
v4.2.7 v3.10.4 v3.8.4
v4.2.9
v4.2.7 v3.10.12 v3.8.4
v4.3.13
v4.1.23
v4.3.13
v4.1.23
DRAGEN Methylation
v4.3.13
v4.1.23
v4.3.13
v4.1.23
DRAGEN Single Cell RNA
v4.2.7 v3.10.4 v3.8.4 v3.7.4
v4.2.7 v3.10.12 v3.8.4 v3.7.4
DRAGEN Somatic
v4.3.13
v4.1.23
v4.3.13
v4.1.23
DRAGEN for DNA Prep with Enrichment
DRAGEN cfDNA Prep with Enrichment
v4.0.3
DRAGEN Protein Quantification
v1.8.33
v1.7.80
NanoString GeoMx NGS Pipeline
v2.0.21
v2.0.21
DRAGEN COVIDSeq Test
v1.3.0
DRAGEN Library QC
v4.3.13
v4.3.13
DRAGEN Small WGS
v4.3.13
v4.3.13
DRAGEN Microbial Enrichment Plus
v4.3.13
v4.3.13
Pipeline
NextSeq 1000/2000
NextSeq 1000/2000
NovaSeq X Series
NovaSeq X Series
NovaSeq 6000 Dx (RUO mode)
NovaSeq 6000 Dx (RUO mode)
NovaSeq 6000
NextSeq 500/550/550 Dx (RUO mode)
MiSeq i100
MiSeq i100
MiSeq
MiniSeq
iSeq 100
Cloud
Onboard
v2.1.1
v2.1.1
✅ = Supported
🟨 = Supported. Change in definition from prior release. See Release Notes and/or Sample Sheet Definition for each version for details.
Blank = Not supported
✅
✅
✅
✅
✅
DRAGEN BCL Convert
[BCLConvert_Settings]
Optional
OverrideCycles
✅
✅
✅
✅
✅
✅
✅
DRAGEN BCL Convert
[BCLConvert_Settings]
Optional
AdapterRead1
✅
✅
✅
✅
✅
✅
✅
DRAGEN BCL Convert
[BCLConvert_Settings]
Optional
AdapterRead2
✅
✅
✅
✅
✅
✅
✅
DRAGEN BCL Convert
[BCLConvert_Settings]
Optional
FastqCompressionFormat
✅
✅
✅
✅
✅
✅
DRAGEN BCL Convert
[BCLConvert_Settings]
Optional
NoLaneSplitting
✅
DRAGEN BCL Convert
[BCLConvert_Data]
Required
SampleID
✅
✅
✅
✅
✅
✅
✅
DRAGEN BCL Convert
[BCLConvert_Data]
Optional
Index
✅
✅
✅
✅
✅
✅
✅
DRAGEN BCL Convert
[BCLConvert_Data]
Optional
Index2
✅
✅
✅
✅
✅
✅
✅
DRAGEN Germline
[DragenGermline_Settings]
Required
SoftwareVersion
✅
✅
✅
✅
✅
✅
DRAGEN Germline
[DragenGermline_Settings]
Required
ReferenceGenomeDir
✅
✅
✅
✅
✅
✅
DRAGEN Germline
[DragenGermline_Settings]
Optional
MapAlignOutFormat
✅
✅
✅
✅
✅
✅
DRAGEN Germline
[DragenGermline_Settings]
Optional
KeepFastq
✅
✅
✅
✅
✅
DRAGEN Germline
[DragenGermline_Data]
Required
SampleID
✅
✅
✅
✅
✅
✅
DRAGEN Enrichment
[DragenEnrichment_Settings]
Required
SoftwareVersion
✅
✅
✅
✅
✅
✅
DRAGEN Enrichment
[DragenEnrichment_Settings]
Required
ReferenceGenomeDir
✅
✅
✅
✅
✅
✅
DRAGEN Enrichment
[DragenEnrichment_Settings]
Optional
MapAlignOutFormat
✅
✅
✅
✅
✅
✅
DRAGEN Enrichment
[DragenEnrichment_Settings]
Required
BedFile
✅
✅
✅
✅
✅
✅
DRAGEN Enrichment
[DragenEnrichment_Settings]
Required
GermlineOrSomatic
✅
✅
✅
✅
✅
DRAGEN Enrichment
[DragenEnrichment_Settings]
Optional
KeepFastq
✅
✅
✅
✅
✅
DRAGEN Enrichment
[DragenEnrichment_Settings]
Optional
AuxNoiseBaselineFile
✅
✅
✅
✅
DRAGEN Enrichment
[DragenEnrichment_Settings]
Optional
AuxCnvPanelOfNormalsFile
✅
✅
DRAGEN Enrichment
[DragenEnrichment_Data]
Required
SampleID
✅
✅
✅
✅
✅
✅
DRAGEN RNA
[DragenRna_Settings]
Required
SoftwareVersion
✅
✅
✅
✅
✅
✅
DRAGEN RNA
DragenRna_Settings]
Required
ReferenceGenomeDir
✅
✅
✅
✅
✅
✅
DRAGEN RNA
DragenRna_Settings]
Optional
RnaGeneAnnotationFile
✅
✅
✅
✅
✅
✅
DRAGEN RNA
DragenRna_Settings]
Optional
MapAlignOutFormat
✅
✅
✅
✅
✅
✅
DRAGEN RNA
DragenRna_Settings]
Optional
KeepFastq
✅
✅
✅
✅
✅
DRAGEN RNA
DragenRna_Settings]
Optional
DifferentialExpressionEnable
✅
✅
✅
✅
DRAGEN RNA
[DragenRna_Data]
Required
SampleID
✅
✅
✅
✅
✅
✅
DRAGEN RNA
[DragenRna_Data]
Optional
Comparison1
✅
✅
✅
✅
DRAGEN RNA
[DragenRna_Data]
Optional
Comparison2
✅
✅
✅
✅
DRAGEN RNA
[DragenRna_Data]
Optional
Comparison3
✅
✅
✅
✅
DRAGEN RNA
[DragenRna_Data]
Optional
Comparison4
✅
✅
✅
✅
DRAGEN RNA
[DragenRna_Data]
Optional
Comparison5
✅
✅
✅
✅
DRAGEN Single Cell
[DragenSingleCellRna_Settings]
Required
SoftwareVersion
✅
✅
✅
✅
✅
DRAGEN Single Cell
[DragenSingleCellRna_Settings]
Required
ReferenceGenomeDir
✅
✅
✅
✅
✅
DRAGEN Single Cell
[DragenSingleCellRna_Settings]
Optional
MapAlignOutFormat
✅
✅
✅
✅
✅
DRAGEN Single Cell
[DragenSingleCellRna_Settings]
Optional
RnaGeneAnnotationFile
✅
✅
✅
✅
✅
DRAGEN Single Cell
[DragenSingleCellRna_Settings]
Optional
KeepFastq
✅
✅
✅
✅
✅
DRAGEN Single Cell
[DragenSingleCellRna_Settings]
Required
BarcodePosition
✅
✅
✅
✅
✅
DRAGEN Single Cell
[DragenSingleCellRna_Settings]
Required
UmiPosition
✅
✅
✅
✅
✅
DRAGEN Single Cell
[DragenSingleCellRna_Settings]
Required
BarcodeRead
✅
✅
✅
✅
✅
DRAGEN Single Cell
[DragenSingleCellRna_Settings]
Required
BarcodeSequenceWhitelist
✅
DRAGEN Single Cell
[DragenSingleCellRna_Settings]
Required
BarcodeSequenceList
✅
✅
✅
✅
DRAGEN Single Cell
[DragenSingleCellRna_Settings]
Required
RnaLibraryType
✅
✅
✅
✅
✅
DRAGEN Single Cell
[DragenSingleCellRna_Data]
Required
SampleID
✅
✅
✅
✅
✅
DRAGEN Amplicon
[DragenAmplicon_Settings]
Required
SoftwareVersion
✅
✅
✅
✅
DRAGEN Amplicon
[DragenAmplicon_Settings]
Required
ReferenceGenomeDir
✅
✅
✅
✅
DRAGEN Amplicon
[DragenAmplicon_Settings]
Required
DnaBedFile
✅
✅
✅
✅
DRAGEN Amplicon
[DragenAmplicon_Settings]
Optional
MapAlignOutFormat
✅
✅
✅
✅
DRAGEN Amplicon
[DragenAmplicon_Settings]
Optional
KeepFastq
✅
✅
✅
✅
DRAGEN Amplicon
[DragenAmplicon_Settings]
Required
DnaGermlineOrSomatic
✅
✅
✅
✅
DRAGEN Amplicon
[DragenAmplicon_Data]
Required
DnaOrRna
✅
✅
✅
✅
DRAGEN Amplicon
[DragenAmplicon_Data]
Required
SampleID
✅
✅
✅
✅
Nanostring
[DragenGeoMxNGS_Settings]
Required
SoftwareVersion
✅
✅
Nanostring
[DragenGeoMxNGS_Settings]
Optional
KeepFastq
✅
✅
Nanostring
[DragenGeoMxNGS_Settings]
Required
GeoMxConfigFile
✅
✅
Nanostring
[DragenGeoMxNGS_Settings]
Required
BarcodeSequenceList
✅
✅
Nanostring
[DragenGeoMxNGS_Data]
Required
SampleID
✅
✅
DRAGEN BCL Convert
[BCLConvert_Settings]
Required
SoftwareVersion
✅
✅
✅ = Supported
🟨 = Supported. Change in definition from prior release. See Release Notes and/or Sample Sheet Definition for each version for details.
Blank = Not supported
[BCLConvert_Settings]
Required
AppVersion
✅
✅
✅
✅
DRAGEN BCL Convert
[BCLConvert_Settings]
Required
FastqCompressionFormat
✅
✅
✅
✅
DRAGEN BCL Convert
[BCLConvert_Settings]
Optional
CreateFastqForIndexReads
✅
✅
✅
✅
DRAGEN BCL Convert
[BCLConvert_Settings]
Optional
TrimUMI
✅
✅
✅
✅
DRAGEN BCL Convert
[BCLConvert_Settings]
Import Option
AdapterBehavior
✅
DRAGEN BCL Convert
[BCLConvert_Settings]
Import Option
AdapterStringency
✅
DRAGEN BCL Convert
[BCLConvert_Settings]
Import Option
MinimumTrimmedReadLength
✅
DRAGEN BCL Convert
[BCLConvert_Settings]
Import Option
MinimumAdapterOverlap
✅
DRAGEN BCL Convert
[BCLConvert_Settings]
Import Option
MaskShortReads
✅
DRAGEN BCL Convert
[BCLConvert_Settings]
Import Option
NoLaneSplitting
✅
DRAGEN BCL Convert
[BCLConvert_Settings]
Import Option
FindAdaptersWithIndels
✅
DRAGEN BCL Convert
[BCLConvert_Settings]
Import Option
IndependentIndexCollisionCheck
✅
DRAGEN BCL Convert
[BCLConvert_Settings]
Import Option
GenerateFastqcMetrics
✅
DRAGEN BCL Convert
[BCLConvert_Data]
Required
SampleID
✅
✅
✅
✅
DRAGEN BCL Convert
[BCLConvert_Data]
Optional
Lane
✅
✅
✅
✅
DRAGEN BCL Convert
[BCLConvert_Data]
Optional
Index
✅
✅
✅
✅
DRAGEN BCL Convert
[BCLConvert_Data]
Optional
Index2
✅
✅
✅
✅
DRAGEN BCL Convert
[BCLConvert_Data]
Optional
OverrideCycles
✅
✅
✅
✅
DRAGEN BCL Convert
[BCLConvert_Data]
Optional
AdapterRead1
✅
✅
✅
✅
DRAGEN BCL Convert
[BCLConvert_Data]
Optional
AdapterRead2
✅
✅
✅
✅
DRAGEN BCL Convert
[BCLConvert_Data]
Optional
BarcodeMismatchesIndex1
✅
✅
✅
✅
DRAGEN BCL Convert
[BCLConvert_Data]
Optional
BarcodeMismatchesIndex1
✅
✅
✅
✅
DRAGEN BCL Convert
[BCLConvert_Data]
Optional
RGID
✅
✅
DRAGEN BCL Convert
[BCLConvert_Data]
Optional
RGPU
✅
✅
DRAGEN BCL Convert
[BCLConvert_Data]
Optional
RGPL
✅
✅
DRAGEN BCL Convert
[BCLConvert_Data]
Optional
RGLB
✅
✅
DRAGEN BCL Convert
[BCLConvert_Data]
Optional
RGCN
✅
✅
DRAGEN BCL Convert
[BCLConvert_Data]
Optional
RGPM
✅
✅
DRAGEN BCL Convert
[BCLConvert_Data]
Import Option
Sample_Project
✅
✅
DRAGEN Germline
[DragenGermline_Settings]
Required
SoftwareVersion
✅
✅
✅
✅
DRAGEN Germline
[DragenGermline_Settings]
Required
AppVersion
✅
✅
✅
✅
DRAGEN Germline
[DragenGermline_Settings]
Required
KeepFastq
✅
✅
✅
✅
DRAGEN Germline
[DragenGermline_Settings]
Required
MapAlignOutFormat
✅
✅
✅
✅
DRAGEN Germline
[DragenGermline_Data]
Required
SampleID
✅
✅
✅
✅
DRAGEN Germline
[DragenGermline_Data]
Required
ReferenceGenomeDir
✅
✅
✅
✅
DRAGEN Germline
[DragenGermline_Data]
Required
VariantCallingMode
✅
🟨
🟨
🟨
DRAGEN Germline
[DragenGermline_Data]
Required
QcCoverage1BedFile
✅
✅
DRAGEN Germline
[DragenGermline_Data]
Required
QcCoverage2BedFile
✅
✅
DRAGEN Germline
[DragenGermline_Data]
Required
QcCoverage3BedFile
✅
✅
DRAGEN Germline
[DragenGermline_Data]
Required
QcCrossContaminationVcfFile
✅
✅
DRAGEN Enrichment
[DragenEnrichment_Settings]
Required
SoftwareVersion
✅
✅
✅
✅
DRAGEN Enrichment
[DragenEnrichment_Settings]
Required
AppVersion
✅
✅
✅
✅
DRAGEN Enrichment
[DragenEnrichment_Settings]
Required
KeepFastq
✅
✅
✅
✅
DRAGEN Enrichment
[DragenEnrichment_Settings]
Optional
MapAlignOutFormat
✅
✅
✅
✅
DRAGEN Enrichment
[DragenEnrichment_Data]
Required
SampleID
✅
✅
✅
✅
DRAGEN Enrichment
[DragenEnrichment_Data]
Required
ReferenceGenomeDir
✅
✅
✅
✅
DRAGEN Enrichment
[DragenEnrichment_Data]
Conditionally Required
BedFile
✅
✅
✅
✅
DRAGEN Enrichment
[DragenEnrichment_Data]
Required
GermlineOrSomatic
✅
✅
✅
✅
DRAGEN Enrichment
[DragenEnrichment_Data]
Optional
AuxNoiseBaselineFile
✅
✅
✅
✅
DRAGEN Enrichment
[DragenEnrichment_Data]
Optional
AuxCnvPanelOfNormalsFile
✅
✅
✅
✅
DRAGEN Enrichment
[DragenEnrichment_Data]
Required
VariantCallingMode
✅
✅
✅
🟨
DRAGEN Enrichment
[DragenEnrichment_Data]
Optional
AuxCnvPopBAlleleVcfFile
✅
DRAGEN Enrichment
[DragenEnrichment_Data]
Optional
AuxGermlineTaggingFile
✅
DRAGEN RNA
[DragenRna_Settings]
Required
SoftwareVersion
✅
✅
✅
✅
DRAGEN RNA
[DragenRna_Settings]
Required
AppVersion
✅
✅
✅
✅
DRAGEN RNA
[DragenRna_Settings]
Required
MapAlignOutFormat
✅
✅
✅
✅
DRAGEN RNA
[DragenRna_Settings]
Required
KeepFastq
✅
✅
✅
✅
DRAGEN RNA
[DragenRna_Settings]
Optional
DifferentialExpressionEnable
✅
✅
✅
✅
DRAGEN RNA
[DragenRna_Data]
Required
SampleID
✅
✅
✅
✅
DRAGEN RNA
[DragenRna_Data]
Required
ReferenceGenomeDir
✅
✅
✅
✅
DRAGEN RNA
[DragenRna_Data]
Optional
RnaGeneAnnotationFile
✅
✅
✅
✅
DRAGEN RNA
[DragenRna_Data]
Required
RnaPipelineMode
✅
✅
✅
✅
DRAGEN RNA
[DragenRna_Data]
Optional
DownSampleNumReads
✅
✅
✅
✅
DRAGEN RNA
[DragenRna_Data]
Optional
Comparison1
✅
✅
✅
✅
DRAGEN RNA
[DragenRna_Data]
Optional
Comparison2
✅
✅
✅
✅
DRAGEN RNA
[DragenRna_Data]
Optional
Comparison3
✅
✅
✅
✅
DRAGEN RNA
[DragenRna_Data]
Optional
Comparison4
✅
✅
✅
✅
DRAGEN RNA
[DragenRna_Data]
Optional
Comparison5
✅
✅
✅
✅
DRAGEN Somatic
[DragenSomatic_Settings]
Required
SoftwareVersion
✅
✅
DRAGEN Somatic
[DragenSomatic_Settings]
Required
AppVersion
✅
✅
DRAGEN Somatic
[DragenSomatic_Settings]
Required
MapAlignOutFormat
✅
✅
DRAGEN Somatic
[DragenSomatic_Settings]
Required
KeepFastq
✅
✅
DRAGEN Somatic
[DragenSomatic_Data]
Optional
AuxNoiseBaselineFile
✅
✅
DRAGEN Somatic
[DragenSomatic_Data]
Optional
AuxSvNoiseBaselineFile
✅
✅
DRAGEN Somatic
[DragenSomatic_Data]
Optional
AuxCnvPopBAlleleVcfFile
✅
✅
DRAGEN Somatic
[DragenSomatic_Data]
Optional
AuxGermlineTaggingFile
✅
✅
DRAGEN Somatic
[DragenSomatic_Data]
Required
VariantCallingMode
✅
✅
DRAGEN Somatic
[DragenSomatic_Data]
Required
Sample_ID
✅
✅
DRAGEN Methylation
[DragenMethylation_Settings]
Required
SoftwareVersion
✅
✅
DRAGEN Methylation
[DragenMethylation_Settings]
Required
AppVersion
✅
✅
DRAGEN Methylation
[DragenMethylation_Settings]
Required
MapAlignOutFormat
✅
✅
DRAGEN Methylation
[DragenMethylation_Settings]
Required
KeepFastq
✅
✅
DRAGEN Methylation
[DragenMethylation_Settings]
Required
UsesTaps
✅
✅
DRAGEN Methylation
[DragenMethylation_Data]
Required
ReferenceGenomeDir
✅
✅
DRAGEN Methylation
[DragenMethylation_Data]
Required
MethylationProtocol
✅
✅
DRAGEN Methylation
[DragenMethylation_Data]
Required
Sample_ID
✅
✅
DRAGEN BCL Convert
[BCLConvert_Settings]
Required
SoftwareVersion
✅
✅
✅
✅
DRAGEN BCL Convert
¹ Depending on the run or library prep kit, this parameter might be required.
¹ Depending on the run or library prep kit, this parameter might be required.
For NovaSeq X Series, there is a limit of four application sections for onboard analysis or eight application sections for cloud analysis.
BCLConvert_Settings
DragenGermline_Settings
DragenEnrichment_Settings
DragenRna_Settings
DragenSomatic_Settings
DragenMethylation_Settings
The run description can contain 255 alphanumeric characters, spaces, dashes, and underscores.
String with ASCII characters except for * and the control characters CR and LF.
RunName
Yes
The run name can contain 255 alphanumeric characters, spaces, dashes, and underscores.
String with ASCII characters except for * and the control characters CR and LF.
Read2Cycles
Yes
Number of cycles for Read 2. Required only when running a paired-end sequencing run.
Must be an integer ≥ 0.
Warning if values in range [1–25] inclusive.
If OverrideCycles is present in the BCL_Settings section, must be consistent with the sum of the Read2 section of OverrideCycles.
CustomRead2Primer
No¹
Indicates if a Custom Read 2 primer is used for the run.
Values true and false allowed. Value true only allowed if Read2Cycles is specified.
LibraryPrepKits
Yes
Identifies the library prep kit used for the run.
String with ASCII characters except for * and the control characters CR and LF.
If more than one library prep kit is being used, use semicolons to separate the names of the different library prep kits.
CreateFastqForIndexReads
No
See
TrimUMI
No
See
AdapterBehavior
No
Import option only. See
AdapterStringency
No
Import option only. See
MinimumTrimmedReadLength
No
Import option only. See
MinimumAdapterOverlap
No
Import option only. See
MaskShortReads
No
Import option only. See
NoLaneSplitting
No
Import option only. See
FindAdaptersWithIndels
No
Import option only. See
IndependentIndexCollisionCheck
No
Import option only. See
BCLConvert_Data
AdapterRead1
No
The sequence of the Read 1 adapter to be masked or trimmed. To trim multiple adapters, separate the sequences with a plus sign (+) indicating independent adapters that must be independently assessed for masking or trimming for each read. Characters must be A, C, G, or T.
A value of na (case insensitive) must be used if:
the AdapterRead1 field is placed in the Data section of the Sample Sheet, and
no adapter trimming is desired for the sample
AdapterRead2
No
See description of AdapterRead1, applied to AdapterRead2.
BarcodeMismatchesIndex1
No
Specifies barcode mismatch tolerance for Index 1. Possible values are 0, 1, or 2, or na. The default value is 1. Only allowed if Index is specified in RunInfo.xml file and in the Reads section of the Sample Sheet.
A value of na must be used if:
BarcodeMismatchesIndex1 exists in the Data section of the Sample Sheet, and
OverrideCycles exist in the data section, and
BarcodeMismatchesIndex2
No
BCLConvert_Settings
SoftwareVersion
Yes
The version of the software to be used to perform BCL conversion on the Sample_ID's that only exist in the BCL Convert Data section of the sample sheet., The version is specified using all three integers included in the DRAGEN version name. For example, 1.0.0.
FastqCompressionFormat
Yes
The compression format for the FASTQ output files. Allowed values are gzip or dragen.
BCLConvert_Data
BCLConvert_Settings
SoftwareVersion
Yes
The version of the software to be used to perform BCL conversion on the Sample_ID's that only exist in the BCL Convert Data section of the sample sheet., The version is specified using all three integers included in the DRAGEN version name. For example, 1.0.0.
FastqCompressionFormat
Yes
The compression format for the FASTQ output files. Allowed values are gzip or dragen.
BCLConvert_Data
BCLConvert_Settings
SoftwareVersion
Yes
The version of the software to be used to perform BCL conversion on the Sample_ID's that only exist in the BCL Convert Data section of the sample sheet., The version is specified using all three integers included in the DRAGEN version name. For example, 1.0.0.
FastqCompressionFormat
Yes
The compression format for the FASTQ output files. Allowed values are gzip or dragen.
GenerateFastqcMetrics
No
Import option only. Enable/disable generation of FAST QC Metrics. Default = True.
BCLConvert_Data
MapAlignOutFormat
Yes
Formatting of the output files. Accepted values are bam, cram, or none. Selecting none produces no map/align output. If MapAlignOutFormat is None, VariantCallingMode cannot be None for any sample
DragenGermline_Data
ReferenceGenomeDir
Yes
Genome name consisting of alphanumeric string with "_" or "-" or ".".
VariantCallingMode
Yes
Variant calling mode for the run.
Accepted values are None, SmallVariantCaller, AllVariantCallers. The option for all variant callers includes Small, Structural, CNV, Repeat Expansions, ROH, CYP2D6, CYP2B6, CYP21A2, SMN, GBA, LPA, RH, and SMN (silent carrier).
QcCoverage1BedFile
No
File name in text (*.txt) or gzip (*.gz) format. Must include the prefix DragenGermline/. Alphanumeric string with underscores (_) or dashes (-) or periods (.) with no spaces allowed. If QcCoverage1BedFile exists in the Data section, but a file is not provided, a value of na must be specified.
QcCoverage2BedFile
No
DragenGermline_Settings
SoftwareVersion
Yes
The version of the DRAGEN software, used to process the DragenGermline pipeline, including conversion to FASTQ, Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
AppVersion
Yes
The version of the workflow-specific application (i.e., DRAGEN Germline), using all three integers included in the version name. For example, 1.0.0.
KeepFastQ
Yes
Select whether FASTQs are saved (true) or discarded (false).
DragenGermline_Data
DragenGermline_Settings
SoftwareVersion
Yes
The version of the DRAGEN software, used to process the DragenGermline pipeline, including conversion to FASTQ, Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
AppVersion
Yes
The version of the workflow-specific application (i.e., DRAGEN Germline), using all three integers included in the version name. For example, 1.0.0.
KeepFastQ
Yes
Select whether FASTQs are saved (true) or discarded (false).
DragenGermline_Data
DragenGermline_Settings
SoftwareVersion
Yes
The version of the DRAGEN software, used to process the DragenGermline pipeline, including conversion to FASTQ, Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
AppVersion
Yes
The version of the workflow-specific application (i.e., DRAGEN Germline), using all three integers included in the version name. For example, 1.0.0.
KeepFastQ
Yes
Select whether FASTQs are saved (true) or discarded (false).
DragenGermline_Data
MapAlignOutFormat
Yes
Formatting of the output files. Accepted values are bam, cram, or none. Selecting none produces no map/align output. If MapAlignOutFormat is None, VariantCallingMode cannot be None for any sample
DragenEnrichment_Data
ReferenceGenomeDir
Yes
Genome name consisting of alphanumeric string with "_" or "-" or ".".
Bedfile
Conditionally required
BED file to be used for analysis in text (*.txt) or gzip (*.gz) format. Only required if VariantCallingMode is not None. Must include the prefix DragenEnrichment/ before the BED file name. Alphanumeric string with _ or - or . with no spaces allowed. The value must be na if BedFile is placed in the Data section and VariantCallingMode = None.
GermlineOrSomatic
Yes
Accepted values are germline or somatic.
AuxNoiseBaselineFile
No
DragenEnrichment_Settings
SoftwareVersion
Yes
The version of the DRAGEN software used to process the DragenEnrichment pipeline, including conversion to FASTQ, , Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
AppVersion
Yes
The version of the workflow-specific application (i.e., DRAGEN Enrichment), using all three integers included in the version name. For example, 1.0.0.
KeepFastQ
Yes
Select whether FASTQs are saved (true) or discarded (false).
DragenEnrichment_Data
DragenEnrichment_Settings
SoftwareVersion
Yes
The version of the DRAGEN software used to process the DragenEnrichment pipeline, including conversion to FASTQ, , Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
AppVersion
Yes
The version of the workflow-specific application (i.e., DRAGEN Enrichment), using all three integers included in the version name. For example, 1.0.0.
KeepFastQ
Yes
Select whether FASTQs are saved (true) or discarded (false).
DragenEnrichment_Data
DragenEnrichment_Settings
SoftwareVersion
Yes
The version of the DRAGEN software used to process the DragenEnrichment pipeline, including conversion to FASTQ, , Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
AppVersion
Yes
The version of the workflow-specific application (i.e., DRAGEN Enrichment), using all three integers included in the version name. For example, 1.0.0.
KeepFastQ
Yes
Select whether FASTQs are saved (true) or discarded (false).
DragenEnrichment_Data
KeepFastQ
Yes
Select whether FASTQs are saved (true) or discarded (false).
DifferentialExpressionEnable
No
Accepted values are true or false. If DifferentialExpressionEnable is true, then RnaPipelineMode must be FullPipeline
DragenRna_Data
ReferenceGenomeDir
Yes
Genome name consisting of alphanumeric string with "_" or "-" or ".". Can be placed in either settings or data section.
RnaGeneAnnotationFile
No
Genotype reference file. Alphanumeric string with "_" or "-" or "." with no spaces allowed. If DifferentialExpressionEnable is True., the GTF file must be provided by the user or included with the reference genome.
RnaPipelineMode
Yes
Accepted values are MapAlign or FullPipeline. The full pipeline option includes quantification and fusion detection.
DownSampleNumReads
No
DragenRna_Settings
SoftwareVersion
Yes
The version of the DRAGEN software used to process the DragenRna pipeline, including conversion to FASTQ,, Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
AppVersion
Yes
The version of the workflow-specific application (i.e., DRAGEN RNA), using all three integers included in the version name. For example, 1.0.0.
MapAlignOutFormat
Yes
Formatting of the output files. Accepted values are bam, cram, or none. Selecting none produces no map/align output. Selecting none is not allowed if RnaPipelineMode is set to FulPipeline for any sample.
DragenRna_Data
DragenRna_Settings
SoftwareVersion
Yes
The version of the DRAGEN software used to process the DragenRNA pipeline, including conversion to FASTQ,, Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
AppVersion
Yes
The version of the workflow-specific application (i.e., DRAGEN RNA), using all three integers included in the version name. For example, 1.0.0.
MapAlignOutFormat
Yes
Formatting of the output files. Accepted values are bam, cram, or none. Selecting none produces no map/align output. Selecting none is not allowed if RnaPipelineMode is set to FulPipeline for any sample.
DragenRna_Data
DragenRna_Settings
SoftwareVersion
Yes
The version of the DRAGEN software used to process the DragenRNA pipeline, including conversion to FASTQ,, Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
AppVersion
Yes
The version of the workflow-specific application (i.e., DRAGEN RNA), using all three integers included in the version name. For example, 1.0.0.
MapAlignOutFormat
Yes
Formatting of the output files. Accepted values are bam, cram, or none. Selecting none produces no map/align output. Selecting none is not allowed if RnaPipelineMode is set to FulPipeline for any sample.
DragenRna_Data
KeepFastq
Required
Select whether FASTQs are saved (true) or discarded (false).
DragenSomatic_Data
AuxNoiseBaselineFile
Optional
Alphanumeric string with "_" or "-" or ".". and no spaces in text (*.txt) or gzip (*.gz) format. Applicable only if GermlineOrSomatic is Somatic and VariantCallingMode is not None.
The value must be na if AuxNoiseBaselineFile is placed in the Data section and no AuxNoiseBaselineFile is provided for the sample."
AuxSvNoiseBaselineFile
Optional
Alphanumeric string with "_" or "-" or "." and no spaces in text (*.txt) or gzip (*.gz) format. Optional if VariantCallingMode is AllVariantCallers.
The value must be na if AuxSvNoiseBaselineFile is placed in the Data section and no AuxSvNoiseBaselineFile is provided for the sample.
AuxCnvPopBAlleleVcfFile
Optional
Alphanumeric string with "_" or "-" or "." and no spaces in text (*.txt) or gzip (*.gz) format. Optional if VariantCallingMode is AllVariantCallers.
The value must be na if AuxCnvPopBAlleleVcfFile is placed in the Data section and no AuxCnvPopBAlleleVcfFile is provided for the sample. CNV output will only be generated if this file is provided.
AuxGermlineTaggingFile
Optional
DragenSomatic_Settings
SoftwareVersion
Required
The version of the DRAGEN software used to process the DragenSomatic pipeline, including conversion to FASTQ. Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
AppVersion
Required
The version of the workflow-specific application (i.e., DRAGEN Somatic), using all three integers included in the version name. For example, 1.0.0.
MapAlignOutFormat
Required
Formatting of the output files. Accepted values are bam, cram, or none. Selecting none produces no map/align output.
DragenSomatic_Data
KeepFastq
Required
Select whether FASTQs are saved (true) or discarded (false).
UsesTaps
Required
Select whether the TAPS assay, which directly converts methylated C to T, is used (true) or not used (false)
DragenMethylation_Data
ReferenceGenomeDir
Required
Genome name consisting of alphanumeric string with "_" or "-" or ".". Can be placed in either settings or data section.
MethylationProtocol
Required
Select the library protocol for methylation analysis from the set {'directional’, ‘non-directional’, ‘directional-complement’,‘pbat’}
Sample_ID
Required
See description in the BCL Convert section
DragenMethylation_Settings
SoftwareVersion
Required
The version of the DRAGEN software used to process the DragenMethylation pipeline, including conversion to FASTQ. Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
AppVersion
Required
The version of the workflow-specific application (i.e., DRAGEN Methylation), using all three integers included in the version name. For example, 1.0.0.
MapAlignOutFormat
Required
Formatting of the output files. Accepted values are bam, cram, or none. Selecting none produces no map/align output.
DragenMethylation_Data
Custom_*
No
Custom field used to capture run metadata.
String with ASCII characters except for * and the control characters CR and LF.
FileFormatVersion
Yes
Used to identify the sample sheet as a v2 sample sheet. This field must always exist in the header section with a value of 2.
Must always be 2.
InstrumentPlatform
No
Identifies the instrument platform to be used for the run.
For example, enter NovaSeqXSeries for NovaSeq X Series.
String with ASCII characters except for * and the control characters CR and LF.
InstrumentType
No
Identifies the instrument to be used for the run.
For example: if using NovaSeq X, populate the field with NovaSeq X.
String with ASCII characters except for * and the control characters CR and LF.
RunDescription
Index1Cycles
No¹
Number of cycles in Index Read 1. Required if more than one sample is present in sample sheet.
Must be an integer ≥ 0.
Depending on your sequencing system and reagent kit, there can be limitations on the number of cycles in the Index Read.
Warning if values in range [1–5] inclusive.
If there is more than 1 sample per lane, must be > 0.
If OverrideCycles is present in the BCL_Settings section, must be consistent with the sum of the Index1 section of OverrideCycles.
Index2Cycles
No¹
Number of cycles in Index Read 2. Required if using dual indexes for demultiplexing.
Must be an integer ≥ 0.
Depending on your sequencing system and reagent kit, there can be limitations on the number of cycles in the Index Read.
Warning if values in range [1–5] inclusive.
If OverrideCycles is present in the BCL_Settings section, must be consistent with the sum of the Index1 section of OverrideCycles.
Read1Cycles
Yes
Number of cycles for Read 1.
CustomIndex1Primer
No¹
Indicates if a Custom Index 1 primer is used for the run.
Values true and false allowed. Value true only allowed if Index1Cycles is specified.
CustomIndex2Primer
No¹
Indicates if a Custom Index 2 primer is used for the run.
Values true and false allowed. Value true only allowed if Index2Cycles is specified.
CustomRead1Primer
No¹
Indicates if a Custom Read 1 primer is used for the run.
SoftwareVersion
Yes
The version of the software to be used to perform BCL conversion on the Sample_ID's that only exist in the BCL Convert Data section of the sample sheet. The version is specified using all three integers included in the DRAGEN version name. For example, 1.0.0.
FastqCompressionFormat
Yes
The compression format for the FASTQ output files. Allowed values are gzip or dragen.
GenerateFastqcMetrics
No
Enable/disable generation of FAST QC Metrics. Default = False. If included in the Sample Sheet, then all DRAGEN versions (for BCL Convert and non-BCL Convert workflows) must be 4.3.13 or later. Not applicable for the Cloud pipeline mode.
SoftwareVersion
Yes
The version of the DRAGEN software used to process the DragenGermline pipeline, including conversion to FASTQ. Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
AppVersion
Yes
The version of the workflow-specific application (i.e., DRAGEN Germline), using all three integers included in the version name. For example, 1.0.0.
KeepFastQ
Yes
SoftwareVersion
Yes
The version of the DRAGEN software used to process the DragenEnrichment pipeline, including conversion to FASTQ. Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
AppVersion
Yes
The version of the workflow-specific application (i.e., DRAGEN Enrichment), using all three integers included in the version name. For example, 1.0.0.
KeepFastQ
Yes
SoftwareVersion
Yes
The version of the DRAGEN software used to process the DragenRna pipeline, including conversion to FASTQ. Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
AppVersion
Yes
The version of the workflow-specific application (i.e., DRAGEN RNA), using all three integers included in the version name. For example, 1.0.0.
MapAlignOutFormat
Yes
Formatting of the output files. Accepted values are bam, cram, or none. Selecting none produces no map/align output. Selecting none is not allowed if RnaPipelineMode is set to FulPipeline for any sample.
SoftwareVersion
Required
The version of the DRAGEN software used to process the DragenSomatic pipeline, including conversion to FASTQ. Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
AppVersion
Required
The version of the workflow-specific application (i.e., DRAGEN Somatic), using all three integers included in the version name. For example, 1.0.0.
MapAlignOutFormat
Required
SoftwareVersion
Required
The version of the DRAGEN software used to process the DragenMethylation pipeline, including conversion to FASTQ. Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
AppVersion
Required
The version of the workflow-specific application (i.e., DRAGEN Methylation), using all three integers included in the version name. For example, 1.0.0.
MapAlignOutFormat
Required
No
Must be an integer > 0.
Warning if less than 26.
If OverrideCycles is present in the BCL_Settings section, must be consistent with the sum of the Read1 section of OverrideCycles.
Values true and false allowed. Value true only allowed if Read1Cycles is specified.
Select whether FASTQs are saved (true) or discarded (false).
Select whether FASTQs are saved (true) or discarded (false).
Formatting of the output files. Accepted values are bam, cram, or none. Selecting none produces no map/align output.
Formatting of the output files. Accepted values are bam, cram, or none. Selecting none produces no map/align output.
No
Specifies the sequencing and indexing cycles to be used when processing the sequencing data. Must adhere to the following requirements:
• Must be same number of fields (delimited by
semicolon) as sequencing and indexing reads specified in RunInfo.xml or and in the Reads section of the Sample Sheet.
• Indexing reads are specified with I, sequencing reads are specified with Y, UMI cycles are specified with U, and trimmed
reads are specified with N.
• The number of cycles specified for each read must equal the number of cycles specified for that read in the RunInfo.xml file and in the Reads section of the Sample Sheet.
• Only one Y or I sequence can be specified per read.
'I' cycles can only be specified for index reads
'Y' cycles can only be specified for genomic reads
Sample_ID
Yes
ID for the sample with the following requirements: • Must be alphanumeric string with _ or - and no spaces. • Case sensitive (i.e., a Sample Sheet with the samples MySample and mysample is not allowed)
The same Sample_ID may exist on more than one row of the Sample Sheet (e.g., one sample spanning more than one lane)
Undetermined is not allowed as a Sample_ID
Lane
Yes
Specifies FASTQ files only for the samples with the specified lane number.
Must adhere to the following requirements:
Must be an integer
Value must be in the range of lanes specified in RunInfo.xml
Index
Yes
The Index 1 (i7) index sequence. Format is a sequence using ACTG. Required if index cycles are specified for the sample in the OverrideCycles.
Must adhere to the following requirements:
Can only contain A, C, G, or T
Length of string must match number of first index cycles in RunInfo.xml or the number specified in OverrideCycles
A value of na must be used if:
Index2
No
See description of Index, applied to Index2.
No
Specifies the sequencing and indexing cycles to be used when processing the sequencing data. Must adhere to the following requirements:
• Must be same number of fields (delimited by
semicolon) as sequencing and indexing reads specified in RunInfo.xml or and in the Reads section of the Sample Sheet.
• Indexing reads are specified with I, sequencing reads are specified with Y, UMI cycles are specified with U, and trimmed
reads are specified with N.
• The number of cycles specified for each read must equal the number of cycles specified for that read in the RunInfo.xml file and in the Reads section of the Sample Sheet.
• Only one Y or I sequence can be specified per read.
'I' cycles can only be specified for index reads
'Y' cycles can only be specified for genomic reads
Sample_ID
Yes
ID for the sample with the following requirements: • Must be alphanumeric string with _ or - and no spaces. • Case sensitive (i.e., a Sample Sheet with the samples MySample and mysample is not allowed)
The same Sample_ID may exist on more than one row of the Sample Sheet (e.g., one sample spanning more than one lane)
Undetermined is not allowed as a Sample_ID
Lane
Yes
Specifies FASTQ files only for the samples with the specified lane number.
Must adhere to the following requirements:
Must be an integer
Value must be in the range of lanes specified in RunInfo.xml
Index
Yes
The Index 1 (i7) index sequence. Format is a sequence using ACTG. Required if index cycles are specified for the sample in the OverrideCycles.
Must adhere to the following requirements:
Can only contain A, C, G, or T
Length of string must match number of first index cycles in RunInfo.xml or the number specified in OverrideCycles
A value of na must be used if:
Index2
No
See description of Index, applied to Index2.
OverrideCycles
No
Specifies the sequencing and indexing cycles to be used when processing the sequencing data. Must adhere to the following requirements: • Must be same number of fields (delimited by semicolon) as sequencing and indexing reads specified in RunInfo.xml and in the Reads section of the Sample Sheet. • Indexing reads are specified with I, sequencing reads are specified with Y, UMI cycles are specified with U, and trimmed reads are specified with N. • The number of cycles specified for each read must equal the number of cycles specified for that read in the RunInfo.xml file and in the Reads section of the Sample Sheet. • Only one Y or I sequence can be specified per read.
'I' cycles can only be specified for index reads
'Y' cycles can only be specified for genomic reads
Sample_ID
Yes
ID for the sample with the following requirements: • Must be alphanumeric string with _ or - and no spaces. • Case sensitive (i.e., a Sample Sheet with the samples MySample and mysample is not allowed)
The same Sample_ID may exist on more than one row of the Sample Sheet (e.g., one sample spanning more than one lane)
Undetermined is not allowed as a Sample_ID
Lane
Yes
Specifies FASTQ files only for the samples with the specified lane number.
Must adhere to the following requirements:
Must be an integer
Value must be in the range of lanes specified in RunInfo.xml
Index
Yes
The Index 1 (i7) index sequence. Format is a sequence using ACTG. Required if index cycles are specified for the sample in the OverrideCycles.
Must adhere to the following requirements:
Can only contain A, C, G, or T
Length of string must match number of first index cycles in RunInfo.xml or the number specified in OverrideCycles
A value of na must be used if:
Index2
No
See description of Index, applied to Index2.
Sample_Project
No
Import option only. See .
QcCoverage3BedFile
No
File name in text (*.txt) or gzip (*.gz) format. Must include the prefix DragenGermline/. Alphanumeric string with underscores (_) or dashes (-) or periods (.) with no spaces allowed. If QcCoverage3BedFile exists in the Data section, but a file is not provided, a value of na must be specified.
QcCrossContaminationVcfFile
No
File name in text (*.txt) or gzip (*.gz) format. Must include the prefix DragenGermline/. Alphanumeric string with underscores (_) or dashes (-) or periods (.) with no spaces allowed. If QcCrossContaminationVcfFile exists in the Data section, but a file is not provided, a value of na must be specified.
Sample_ID
Yes
See description in the BCL Convert section
AuxCnvPanelOfNormalsFile
No
Alphanumeric string with "_" or "-" or "." and no spaces in text (*.txt) or gzip (*.gz) format. Optional if VariantCallingMode is AllVariantCallers.
The value must be na if AuxCnvPanelOfNormalsFile is placed in the Data section and no AuxCnvPanelOfNormalsFile is provided for the sample.
VariantCallingMode
Yes
Variant calling mode for the run.
Accepted values are None, SmallVariantCaller, AllVariantCallers. The option for all variant callers includes Small, Structural, and CNV callers (if panel of normals is provided).
Sample_ID
Yes
See description in the BCL Convert section
AuxCnvPanelOfNormalsFile
No
Alphanumeric string with "_" or "-" or "." and no spaces in text (*.txt) or gzip (*.gz) format. Optional if VariantCallingMode is AllVariantCallers.
The value must be na if AuxCnvPanelOfNormalsFile is placed in the Data section and no AuxCnvPanelOfNormalsFile is provided for the sample.
VariantCallingMode
Yes
Variant calling mode for the run.
Accepted values are None, SmallVariantCaller, AllVariantCallers. The option for all variant callers includes Small, Structural, and CNV callers (if panel of normals is provided).
Sample_ID
Yes
See description in the BCL Convert section
AuxCnvPanelOfNormalsFile
No
Alphanumeric string with "_" or "-" or "." and no spaces in text (*.txt) or gzip (*.gz) format. Optional if VariantCallingMode is AllVariantCallers.
The value must be na if AuxCnvPanelOfNormalsFile is placed in the Data section and no AuxCnvPanelOfNormalsFile is provided for the sample.
VariantCallingMode
Yes
Variant calling mode for the run.
Accepted values are None, SmallVariantCaller, AllVariantCallers. The option for all variant callers includes Small, Structural, and CNV callers (if panel of normals is provided).
Sample_ID
Yes
See description in the BCL Convert section
Sample_ID
Yes
See description in the BCL Convert section
Comparison1
No
Accepted values are control, comparison, or na. Use only if DifferentialExpressionEnable is true. If RNAPipelineMode is MapAlign, this value must be na. Must have at least 2 control and 2 comparison samples. May have a max of 15 control or comparison samples. All control and comparison samples must have FullPipeline for RnaPipelineMode value and have the same ReferenceGenomeDir and RnaGeneAnnotationFile values.
Comparison2
No
See Comparsion1 description.
Comparison3
No
See Comparsion1 description.
Comparison4
No
See Comparsion1 description.
Comparison5
No
See Comparsion1 description.
Sample_ID
Yes
See description in the BCL Convert section
Comparison1
No
Accepted values are control, comparison, or na. Use only if DifferentialExpressionEnable is true. If RNAPipelineMode is MapAlign, this value must be na. Must have at least 2 control and 2 comparison samples. May have a max of 15 control or comparison samples. All control and comparison samples must have FullPipeline for RnaPipelineMode value and have the same ReferenceGenomeDir and RnaGeneAnnotationFile values.
Comparison2
No
See Comparison1 description.
Comparison3
No
See Comparison1 description.
Comparison4
No
See Comparison1 description.
Comparison5
No
See Comparison1 description.
Sample_ID
Yes
See description in the BCL Convert section
Comparison1
No
Accepted values are control, comparison, or na. Use only if DifferentialExpressionEnable is true. If RNAPipelineMode is MapAlign, this value must be na. Must have at least 2 control and 2 comparison samples. May have a max of 15 control or comparison samples. All control and comparison samples must have FullPipeline for RnaPipelineMode value and have the same ReferenceGenomeDir and RnaGeneAnnotationFile values.
Comparison2
No
See Comparison1 Description.
Comparison3
No
See Comparison1 Description.
Comparison4
No
See Comparison1 Description.
Comparison5
No
See Comparison1 Description.
VariantCallingMode
Required
Variant calling mode for the run.
Accepted values are None, SmallVariantCaller, AllVariantCallers. The option for all variant callers includes Small, Structural, and CNV callers (if panel of normals is provided).
Sample_ID
Required
See description in the BCL Convert section
See description of BarcodeMismatchesIndex1, applied to BarcodeMismatchesIndex2.
OverrideCycles
No
Specifies the sequencing and indexing cycles to be used when processing the sequencing data. Must adhere to the Specifies the sequencing and indexing cycles to be used when processing the sequencing data. Must adhere to the following requirements: • Must be same number of fields (delimited by semicolon) as sequencing and indexing reads specified in RunInfo.xml and in the Reads section of the Sample Sheet. • Indexing reads are specified with I, sequencing reads are specified with Y, UMI cycles are specified with U, and trimmed reads are specified with N. • The number of cycles specified for each read must equal the number of cycles specified for that read in the RunInfo.xml file and in the Reads section of the Sample Sheet. • Only one Y or I sequence can be specified per read.
'I' cycles can only be specified for index reads
'Y' cycles can only be specified for genomic reads
The total number of cycles used for demultiplexing each index cannot exceed 27. Note that this includes all cycles between the first "I" cycle used by any sample and the last "I" cycle used by any sample within each index.
"I5N3;I5N3" : counts as 5 bases toward the limit of 27 for Index1, and as 5 bases toward the limit of 27 for Index2
"I4N1I3;I5N3": counts as 8 bases toward the limit of 27 for Index1, and as 5 bases toward the limit of 27 for Index2
The following are examples of OverrideCycles input: U8Y143;I8;I8;U8Y143 N10Y66;I6;N10Y66
For a sample sheet containing two samples having the following OverrideCycles:
Y151; I8N2; N10; Y151
Y151; N2I8; I8N2; Y151
the number of cycles used for demultiplexing sums to 18.
For sequencers with RunInfo.xml having "IsReverseComplement" set to "Y" for Index2, then Index2 is processed in reverse complement orientation. Any trimming should be applied to the end of the sequence as it transitions to the adapter, which means the "N" cycles specified in OverrideCycles must be in the beginning of the index read.
Example of 8bp i5 index with the last two (adapter) bases to be trimmed:
Forward i5: XXATCGCGGT
ReverseComp i5: ACCGCGATXX (this is the direction of sequencing)
OverrideCycles for Index2: N2I8
where XX is part of the adapter sequence.
Although Index2 is processed on the sequencer in reverse complement format, Index2 and OverrideCycles are entered in the Sample Sheet in forward, non-complemented format for user convenience (see diagram below).
Note that NovaSeq X has a RunInfo.xml with "IsReverseComplement" set to "Y" for Index2.
Sample_ID
Yes
ID for the sample with the following requirements: • Must be alphanumeric string with _ or - and no spaces. • Case sensitive (i.e., a Sample Sheet with the samples MySample and mysample is not allowed)
The same Sample_ID may exist on more than one row of the Sample Sheet (e.g., one sample spanning more than one lane)
Undetermined is not allowed as a Sample_ID
Lane
Yes
Specifies FASTQ files only for the samples with the specified lane number.
Must adhere to the following requirements:
Must be an integer
Value must be in the range of lanes specified in RunInfo.xml
Ranges are not supported with '-' or '+'
If not supplied, it is assumed that all samples are present in all lanes specified in the RunInfo.xml
If supplied, only lanes specified in the column will be converted from BCL to FASTQ
For NovaSeqX, values must be in the range of lanes specified in RunInfo.xml
1.5B Flow Cell: 1-2
10B Flow Cell: 1-8
Index
Yes
The Index 1 (i7) index sequence. Format is a sequence using ACTG. Required if index cycles are specified for the sample in the OverrideCycles.
Must adhere to the following requirements:
Can only contain A, C, G, or T
Length of string must match number of first index cycles in RunInfo.xml or the number specified in OverrideCycles
A value of NA must be used if:
No indexes are specified in OverrideCycles for a sample
Index2
No
See description of Index, applied to Index2.
Sample_Project
No
Import option only. See https://help.dragen.illumina.com/product-guides/dragen-v4.3/bcl-conversion. "Logs" and "Reports" are invalid values.
AdapterRead1
No
The sequence of the Read 1 adapter to be masked or trimmed. To trim multiple adapters, separate the sequences with a plus sign (+) indicating independent adapters that must be independently assessed for masking or trimming for each read. Characters must be A, C, G, or T.
A value of na (case insensitive) must be used if:
the AdapterRead1 field is placed in the Data section of the Sample Sheet, and
no adapter trimming is desired for the sample
AdapterRead2
No
See description of AdapterRead1, applied to AdapterRead2.
BarcodeMismatchesIndex1
No
Specifies barcode mismatch tolerance for Index 1. Possible values are 0, 1, or 2, or na. The default value is 1. Only allowed if Index is specified in RunInfo.xml file and in the Reads section of the Sample Sheet.
A value of na must be used if:
BarcodeMismatchesIndex1 exists in the Data section of the Sample Sheet, and
OverrideCycles exist in the data section, and
Index 1 is masked out for the sample in the OverrideCycles
BarcodeMismatchesIndex2
No
See description of BarcodeMismatchesIndex1, applied to BarcodeMismatchesIndex2.
AdapterRead1
No
The sequence of the Read 1 adapter to be masked or trimmed. To trim multiple adapters, separate the sequences with a plus sign (+) indicating independent adapters that must be independently assessed for masking or trimming for each read. Characters must be A, C, G, or T.
A value of na (case insensitive) must be used if:
the AdapterRead1 field is placed in the Data section of the Sample Sheet, and
no adapter trimming is desired for the sample
AdapterRead2
No
See description of AdapterRead1, applied to AdapterRead2.
BarcodeMismatchesIndex1
No
Specifies barcode mismatch tolerance for Index 1. Possible values are 0, 1, or 2, or na. The default value is 1. Only allowed if Index is specified in RunInfo.xml file and in the Reads section of the Sample Sheet.
A value of na must be used if:
BarcodeMismatchesIndex1 exists in the Data section of the Sample Sheet, and
OverrideCycles exist in the data section, and
Index 1 is masked out for the sample in the OverrideCycles
BarcodeMismatchesIndex2
No
See description of BarcodeMismatchesIndex1, applied to BarcodeMismatchesIndex2.
CreateFastqForIndexReads
No
TrimUMI
No
AdapterBehavior
No
Import option only. See https://help.dragen.illumina.com/product-guides/dragen-v4.3/bcl-conversion
AdapterStringency
No
Import option only. See https://help.dragen.illumina.com/product-guides/dragen-v4.3/bcl-conversion
MinimumTrimmedReadLength
No
Import option only. See https://help.dragen.illumina.com/product-guides/dragen-v4.3/bcl-conversion
MinimumAdapterOverlap
No
Import option only. See https://help.dragen.illumina.com/product-guides/dragen-v4.3/bcl-conversion
MaskShortReads
No
Import option only. See https://help.dragen.illumina.com/product-guides/dragen-v4.3/bcl-conversion
NoLaneSplitting
No
Import option only. See https://help.dragen.illumina.com/product-guides/dragen-v4.3/bcl-conversion
FindAdaptersWithIndels
No
Import option only. See https://help.dragen.illumina.com/product-guides/dragen-v4.3/bcl-conversion
IndependentIndexCollisionCheck
No
Import option only. See https://help.dragen.illumina.com/product-guides/dragen-v4.3/bcl-conversion
AdapterRead1
No
The sequence of the Read 1 adapter to be masked or trimmed. To trim multiple adapters, separate the sequences with a plus sign (+) indicating independent adapters that must be independently assessed for masking or trimming for each read. Characters must be A, C, G, or T.
A value of na (case insensitive) must be used if:
the AdapterRead1 field is placed in the Data section of the Sample Sheet, and
no adapter trimming is desired for the sample
AdapterRead2
No
See description of AdapterRead1, applied to AdapterRead2.
BarcodeMismatchesIndex1
No
Specifies barcode mismatch tolerance for Index 1. Possible values are 0, 1, or 2, or na. The default value is 1. Only allowed if Index is specified in RunInfo.xml file and in the Reads section of the Sample Sheet.
A value of na must be used if:
BarcodeMismatchesIndex1 exists in the Data section of the Sample Sheet, and
OverrideCycles exist in the data section, and
Index 1 is masked out for the sample in the OverrideCycles
BarcodeMismatchesIndex2
No
File name in text (*.txt) or gzip (*.gz) format. Must include the prefix DragenGermline/. Alphanumeric string with underscores (_) or dashes (-) or periods (.) with no spaces allowed. If QcCoverage2BedFile exists in the Data section, but a file is not provided, a value of na must be specified.
QcCoverage3BedFile
No
File name in text (*.txt) or gzip (*.gz) format. Must include the prefix DragenGermline/. Alphanumeric string with underscores (_) or dashes (-) or periods (.) with no spaces allowed. If QcCoverage3BedFile exists in the Data section, but a file is not provided, a value of na must be specified.
QcCrossContaminationVcfFile
No
File name in text (*.txt) or gzip (*.gz) format. Must include the prefix DragenGermline/. Alphanumeric string with underscores (_) or dashes (-) or periods (.) with no spaces allowed. If QcCrossContaminationVcfFile exists in the Data section, but a file is not provided, a value of na must be specified.
Sample_ID
Yes
See description in the BCL Convert section
MapAlignOutFormat
Yes
Formatting of the output files. Accepted values are bam, cram, or none. Selecting none produces no map/align output. If MapAlignOutFormat is None, VariantCallingMode cannot be None for any sample
ReferenceGenomeDir
Yes
Genome name consisting of alphanumeric string with "_" or "-" or ".".
VariantCallingMode
Yes
Variant calling mode for the run.
Accepted values are None, SmallVariantCaller, AllVariantCallers. The option for all variant callers includes Small, Structural, CNV, Repeat Expansions, ROH, CYP2D6, CYP2B6, CYP21A2, SMN, and GBA.
QcCoverage1BedFile
No
File name in text (*.txt) or gzip (*.gz) format. Must include the prefix DragenGermline/. Alphanumeric string with underscores (_) or dashes (-) or periods (.) with no spaces allowed. If QcCoverage1BedFile exists in the Data section, but a file is not provided, a value of na must be specified.
QcCoverage2BedFile
No
MapAlignOutFormat
Yes
Formatting of the output files. Accepted values are bam, cram, or none. Selecting none produces no map/align output. If MapAlignOutFormat is None, VariantCallingMode cannot be None for any sample
ReferenceGenomeDir
Yes
Genome name consisting of alphanumeric string with "_" or "-" or ".".
VariantCallingMode
Yes
Variant calling mode for the run.
Accepted values are None, SmallVariantCaller, AllVariantCallers. The option for all variant callers includes Small, Structural, CNV, Repeat Expansions, ROH, CYP2D6, CYP2B6, CYP21A2, SMN, and GBA.
Sample_ID
Yes
See description in the BCL Convert section
MapAlignOutFormat
Yes
Formatting of the output files. Accepted values are bam, cram, or none. Selecting none produces no map/align output. If MapAlignOutFormat is None, VariantCallingMode cannot be None for any sample
ReferenceGenomeDir
Yes
Genome name consisting of alphanumeric string with "_" or "-" or ".".
VariantCallingMode
Yes
Variant calling mode for the run.
Accepted values are None, SmallVariantCaller, AllVariantCallers. The option for all variant callers includes Small, Structural, CNV, Repeat Expansions, ROH, CYP2D6.
Sample_ID
Yes
See description in the BCL Convert section
Alphanumeric string with "_" or "-" or ".". and no spaces in text (*.txt) or gzip (*.gz) format. Applicable only if GermlineOrSomatic is Somatic and VariantCallingMode is not None.
The value must be na if AuxNoiseBaselineFile is placed in the Data section and no AuxNoiseBaselineFile is provided for the sample."
AuxCnvPanelOfNormalsFile
No
Alphanumeric string with "_" or "-" or "." and no spaces in text (*.txt) or gzip (*.gz) format. Optional if VariantCallingMode is AllVariantCallers.
The value must be na if AuxCnvPanelOfNormalsFile is placed in the Data section and no AuxCnvPanelOfNormalsFile is provided for the sample.
VariantCallingMode
Yes
Variant calling mode for the run.
Accepted values are None, SmallVariantCaller, AllVariantCallers. The option for all variant callers includes Small, Structural, and CNV callers (if panel of normals is provided).
AuxCnvPopBAlleleVcfFile
No
Alphanumeric string with _ or - or . with no spaces allowed. Text or gzip format. Optional only if VariantCallingMode = AllVariantCallers and GermlineOrSomatic=somatic. Error otherwise. A note should be added to the UI to indicate that CNV output will only be generated if this file is provided.
AuxGermlineTaggingFile
No
Alphanumeric string with _ or - or . with no spaces allowed. Binary file with .bin extension. Optional when SmallVariantCaller or AllVariantCaller option selected and GermlineOrSomatic=somatic. Error otherwise.
Sample_ID
Yes
See description in the BCL Convert section
MapAlignOutFormat
Yes
Formatting of the output files. Accepted values are bam, cram, or none. Selecting none produces no map/align output. If MapAlignOutFormat is None, VariantCallingMode cannot be None for any sample
ReferenceGenomeDir
Yes
Genome name consisting of alphanumeric string with "_" or "-" or ".".
Bedfile
Conditionally required
BED file to be used for analysis in text (*.txt) or gzip (*.gz) format. Only required if VariantCallingMode is not None. Must include the prefix DragenEnrichment/ before the BED file name. Alphanumeric string with _ or - or . with no spaces allowed. The value must be na if BedFile is placed in the Data section and VariantCallingMode = None.
GermlineOrSomatic
Yes
Accepted values are germline or somatic.
AuxNoiseBaselineFile
No
MapAlignOutFormat
Yes
Formatting of the output files. Accepted values are bam, cram, or none. Selecting none produces no map/align output. If MapAlignOutFormat is None, VariantCallingMode cannot be None for any sample
ReferenceGenomeDir
Yes
Genome name consisting of alphanumeric string with "_" or "-" or ".".
Bedfile
Conditionally required
BED file to be used for analysis in text (*.txt) or gzip (*.gz) format. Only required if VariantCallingMode is not None. Must include the prefix DragenEnrichment/ before the BED file name. Alphanumeric string with _ or - or . with no spaces allowed. The value must be na if BedFile is placed in the Data section and VariantCallingMode = None.
GermlineOrSomatic
Yes
Accepted values are germline or somatic.
AuxNoiseBaselineFile
No
MapAlignOutFormat
Yes
Formatting of the output files. Accepted values are bam, cram, or none. Selecting none produces no map/align output. If MapAlignOutFormat is None, VariantCallingMode cannot be None for any sample
ReferenceGenomeDir
Yes
Genome name consisting of alphanumeric string with "_" or "-" or ".".
Bedfile
Conditionally required
BED file to be used for analysis in text (*.txt) or gzip (*.gz) format. Only required if VariantCallingMode is not None. Must include the prefix DragenEnrichment/ before the BED file name. Alphanumeric string with _ or - or . with no spaces allowed. The value must be na if BedFile is placed in the Data section and VariantCallingMode = None.
GermlineOrSomatic
Yes
Accepted values are germline or somatic.
AuxNoiseBaselineFile
No
Specifies the number of fragments to downsample to. For paired-end sequencing, the number of reads at the down-sampling output will be twice the number of fragments specified. Accepted values are integers. If DownSampleNumReads is placed in the Data section, and downsampling is not desired for that sample, a value of na must be used.
Sample_ID
Yes
See description in the BCL Convert section
Comparison1
No
Accepted values are control, comparison, or na. Use only if DifferentialExpressionEnable is true. If RNAPipelineMode is MapAlign, this value must be na. Must have at least 2 control and 2 comparison samples. May have a max of 15 control or comparison samples. All control and comparison samples must have FullPipeline for RnaPipelineMode value and have the same ReferenceGenomeDir and RnaGeneAnnotationFile values.
Comparison2
No
See Comparison1 Description.
Comparison3
No
See Comparison1 Description.
Comparison4
No
See Comparison1 Description.
Comparison5
No
See Comparison1 Description.
KeepFastQ
Yes
Select whether FASTQs are saved (true) or discarded (false).
DifferentialExpressionEnable
No
Accepted values are true or false. If DifferentialExpressionEnable is true, then RnaPipelineMode must be FullPipeline
ReferenceGenomeDir
Yes
Genome name consisting of alphanumeric string with "_" or "-" or ".". Can be placed in either settings or data section.
RnaGeneAnnotationFile
No
Genotype reference file. Alphanumeric string with "_" or "-" or "." with no spaces allowed. If DifferentialExpressionEnable is True., the GTF file must be provided by the user or included with the reference genome.
RnaPipelineMode
Yes
Accepted values are MapAlign or FullPipeline. The full pipeline option includes quantification and fusion detection.
DownSampleNumReads
No
KeepFastQ
Yes
Select whether FASTQs are saved (true) or discarded (false).
DifferentialExpressionEnable
No
Accepted values are true or false. If DifferentialExpressionEnable is true, then RnaPipelineMode must be FullPipeline
ReferenceGenomeDir
Yes
Genome name consisting of alphanumeric string with "_" or "-" or ".". Can be placed in either settings or data section.
RnaGeneAnnotationFile
No
Genotype reference file. Alphanumeric string with "_" or "-" or "." with no spaces allowed. If DifferentialExpressionEnable is True., the GTF file must be provided by the user or included with the reference genome.
RnaPipelineMode
Yes
Accepted values are MapAlign or FullPipeline. The full pipeline option includes quantification and fusion detection.
DownSampleNumReads
No
KeepFastQ
Yes
Select whether FASTQs are saved (true) or discarded (false).
DifferentialExpressionEnable
No
Accepted values are true or false. If DifferentialExpressionEnable is true, then RnaPipelineMode must be FullPipeline
ReferenceGenomeDir
Yes
Genome name consisting of alphanumeric string with "_" or "-" or ".". Can be placed in either settings or data section.
RnaGeneAnnotationFile
No
Genotype reference file. Alphanumeric string with "_" or "-" or "." with no spaces allowed. If DifferentialExpressionEnable is True., the GTF file must be provided by the user or included with the reference genome.
RnaPipelineMode
Yes
Accepted values are MapAlign or FullPipeline. The full pipeline option includes quantification and fusion detection.
DownSampleNumReads
No
Alphanumeric string with "_" or "-" or "." and no spaces in text (*.txt) or gzip (*.gz) format. Optional if VariantCallingMode is AllVariantCallers.
The value must be na if AuxGermlineTaggingFile is placed in the Data section and no AuxGermlineTaggingFile is provided for the sample. Germline tagging output will only be generated if this file is provided.
VariantCallingMode
Required
Variant calling mode for the run.
Accepted values are None, SmallVariantCaller, AllVariantCallers. The option for all variant callers includes Small, Structural, and CNV callers (if panel of normals is provided).
Sample_ID
Required
See description in the BCL Convert section
KeepFastq
Required
Select whether FASTQs are saved (true) or discarded (false).
AuxNoiseBaselineFile
Optional
Alphanumeric string with "_" or "-" or ".". and no spaces in text (*.txt) or gzip (*.gz) format. Applicable only if GermlineOrSomatic is Somatic and VariantCallingMode is not None.
The value must be na if AuxNoiseBaselineFile is placed in the Data section and no AuxNoiseBaselineFile is provided for the sample."
AuxSvNoiseBaselineFile
Optional
Alphanumeric string with "_" or "-" or "." and no spaces in text (*.txt) or gzip (*.gz) format. Optional if VariantCallingMode is AllVariantCallers.
The value must be na if AuxSvNoiseBaselineFile is placed in the Data section and no AuxSvNoiseBaselineFile is provided for the sample.
AuxCnvPopBAlleleVcfFile
Optional
Alphanumeric string with "_" or "-" or "." and no spaces in text (*.txt) or gzip (*.gz) format. Optional if VariantCallingMode is AllVariantCallers.
The value must be na if AuxCnvPopBAlleleVcfFile is placed in the Data section and no AuxCnvPopBAlleleVcfFile is provided for the sample. CNV output will only be generated if this file is provided.
AuxGermlineTaggingFile
Optional
KeepFastq
Required
Select whether FASTQs are saved (true) or discarded (false).
UsesTaps
Required
Select whether the TAPS assay, which directly converts methylated C to T, is used (true) or not used (false)
ReferenceGenomeDir
Required
Genome name consisting of alphanumeric string with "_" or "-" or ".". Can be placed in either settings or data section.
MethylationProtocol
Required
Select the library protocol for methylation analysis from the set {'directional’, ‘non-directional’, ‘directional-complement’,‘pbat’}
Sample_ID
Required
See description in the BCL Convert section
OverrideCycles
OverrideCycles
See description of BarcodeMismatchesIndex1, applied to BarcodeMismatchesIndex2.
File name in text (*.txt) or gzip (*.gz) format. Must include the prefix DragenGermline/. Alphanumeric string with underscores (_) or dashes (-) or periods (.) with no spaces allowed. If QcCoverage2BedFile exists in the Data section, but a file is not provided, a value of na must be specified.
Alphanumeric string with "_" or "-" or ".". and no spaces in text (*.txt) or gzip (*.gz) format. Applicable only if GermlineOrSomatic is Somatic and VariantCallingMode is not None.
The value must be na if AuxNoiseBaselineFile is placed in the Data section and no AuxNoiseBaselineFile is provided for the sample."
Alphanumeric string with "_" or "-" or ".". and no spaces in text (*.txt) or gzip (*.gz) format. Applicable only if GermlineOrSomatic is Somatic and VariantCallingMode is not None.
The value must be na if AuxNoiseBaselineFile is placed in the Data section and no AuxNoiseBaselineFile is provided for the sample."
Alphanumeric string with "_" or "-" or ".". and no spaces in text (*.txt) or gzip (*.gz) format. Applicable only if GermlineOrSomatic is Somatic and VariantCallingMode is not None.
The value must be na if AuxNoiseBaselineFile is placed in the Data section and no AuxNoiseBaselineFile is provided for the sample."
Specifies the number of fragments to downsample to. For paired-end sequencing, the number of reads at the down-sampling output will be twice the number of fragments specified. Accepted values are integers. If DownSampleNumReads is placed in the Data section, and downsampling is not desired for that sample, a value of na must be used.
Specifies the number of fragments to downsample to. For paired-end sequencing, the number of reads at the down-sampling output will be twice the number of fragments specified. Accepted values are integers. If DownSampleNumReads is placed in the Data section, and downsampling is not desired for that sample, a value of na must be used.
Specifies the number of fragments to downsample to. For paired-end sequencing, the number of reads at the down-sampling output will be twice the number of fragments specified. Accepted values are integers. If DownSampleNumReads is placed in the Data section, and downsampling is not desired for that sample, a value of na must be used.
Alphanumeric string with "_" or "-" or "." and no spaces in text (*.txt) or gzip (*.gz) format. Optional if VariantCallingMode is AllVariantCallers.
The value must be na if AuxGermlineTaggingFile is placed in the Data section and no AuxGermlineTaggingFile is provided for the sample. Germline tagging output will only be generated if this file is provided.
The total number of cycles used for demultiplexing by both indexes together cannot exceed 27. Note that this includes all cycles between the first "I" cycle used by any sample and the last "I" cycle used by any sample within each index.
"I5N3;I5N3" : counts as 10 bases toward the limit of 27
"I4N1I3;I5N3": counts as (8+5) 13 bases toward the limit of 27
The following are examples of OverrideCycles input: U8Y143;I8;I8;U8Y143 N10Y66;I6;N10Y66
For a sample sheet containing two samples having the following OverrideCycles
Y151; I8N2; N10; Y151
Y151; N2I8; I8N2; Y151
the number of cycles used for demultiplexing sums to 18.
For sequencers with RunInfo.xml having "IsReverseComplement" set to "Y" for Index2, then Index2 is processed in reverse complement orientation. Any trimming should be applied to the end of the sequence as it transitions to the adapter, which means the "N" cycles specified in OverrideCycles must be in the beginning of the index read.
Example of 8bp i5 index with the last two (adapter) bases to be trimmed:
Forward i5: XXATCGCGGT
ReverseComp i5: ACCGCGATXX (this is the direction of sequencing)
OverrideCycles for Index2: N2I8
where XX is part of the adapter sequence.
Although Index2 is processed on the sequencer in reverse complement format, Index2 and OverrideCycles are entered in the Sample Sheet in forward, non-complemented format for user convenience (see diagram below).
Note that NovaSeq X has a RunInfo.xml with "IsReverseComplement" set to "Y" for Index2.
If not supplied, it is assumed that all samples are present in all lanes specified in the RunInfo.xml
If supplied, only lanes specified in the column will be converted from BCL to FASTQ
For NovaSeqX, values must be in the range of lanes specified in RunInfo.xml
1.5B Flow Cell: 1-2
10B Flow Cell: 1-8
25B Flow Cell: 1-8
No indexes are specified in OverrideCycles for a sample
The total number of cycles used for demultiplexing by both indexes together cannot exceed 27. Note that this includes all cycles between the first "I" cycle used by any sample and the last "I" cycle used by any sample within each index.
"I5N3;I5N3" : counts as 10 bases toward the limit of 27
"I4N1I3;I5N3": counts as (8+5) 13 bases toward the limit of 27
The following are examples of OverrideCycles input: U8Y143;I8;I8;U8Y143 N10Y66;I6;N10Y66
For a sample sheet containing two samples having the following OverrideCycles
Y151; I8N2; N10; Y151
Y151; N2I8; I8N2; Y151
the number of cycles used for demultiplexing sums to 18.
For sequencers with RunInfo.xml having "IsReverseComplement" set to "Y" for Index2, then Index2 is processed in reverse complement orientation. Any trimming should be applied to the end of the sequence as it transitions to the adapter, which means the "N" cycles specified in OverrideCycles must be in the beginning of the index read.
Example of 8bp i5 index with the last two (adapter) bases to be trimmed:
Forward i5: XXATCGCGGT
ReverseComp i5: ACCGCGATXX (this is the direction of sequencing)
OverrideCycles for Index2: N2I8
where XX is part of the adapter sequence.
Although Index2 is processed on the sequencer in reverse complement format, Index2 and OverrideCycles are entered in the Sample Sheet in forward, non-complemented format for user convenience (see diagram below).
Note that NovaSeq X has a RunInfo.xml with "IsReverseComplement" set to "Y" for Index2.
If not supplied, it is assumed that all samples are present in all lanes specified in the RunInfo.xml
If supplied, only lanes specified in the column will be converted from BCL to FASTQ
For NovaSeqX, values must be in the range of lanes specified in RunInfo.xml
1.5B Flow Cell: 1-2
10B Flow Cell: 1-8
25B Flow Cell: 1-8
No indexes are specified in OverrideCycles for a sample
The total number of cycles used for demultiplexing each index cannot exceed 27. Note that this includes all cycles between the first "I" cycle used by any sample and the last "I" cycle used by any sample within each index.
"I5N3;I5N3" : counts as 5 bases toward the limit of 27 for Index1, and as 5 bases toward the limit of 27 for Index2
"I4N1I3;I5N3": counts as 8 bases toward the limit of 27 for Index1, and as 5 bases toward the limit of 27 for Index2
The following are examples of OverrideCycles input: U8Y143;I8;I8;U8Y143 N10Y66;I6;N10Y66
For a sample sheet containing two samples having the following OverrideCycles
Y151; I8N2; N10; Y151
Y151; N2I8; I8N2; Y151
the number of cycles used for demultiplexing sums to 18.
For sequencers with RunInfo.xml having "IsReverseComplement" set to "Y" for Index2, then Index2 is processed in reverse complement orientation. Any trimming should be applied to the end of the sequence as it transitions to the adapter, which means the "N" cycles specified in OverrideCycles must be in the beginning of the index read.
Example of 8bp i5 index with the last two (adapter) bases to be trimmed:
Forward i5: XXATCGCGGT
ReverseComp i5: ACCGCGATXX (this is the direction of sequencing)
OverrideCycles for Index2: N2I8
where XX is part of the adapter sequence.
Although Index2 is processed on the sequencer in reverse complement format, Index2 and OverrideCycles are entered in the Sample Sheet in forward, non-complemented format for user convenience (see diagram below).
Note that NovaSeq X has a RunInfo.xml with "IsReverseComplement" set to "Y" for Index2.
If not supplied, it is assumed that all samples are present in all lanes specified in the RunInfo.xml
If supplied, only lanes specified in the column will be converted from BCL to FASTQ
For NovaSeqX, values must be in the range of lanes specified in RunInfo.xml
1.5B Flow Cell: 1-2
10B Flow Cell: 1-8
25B Flow Cell: 1-8
No indexes are specified in OverrideCycles for a sample
¹ Depending on the run or library prep kit, this parameter might be required.
BCLConvert_Settings
DragenGermline_Settings
DragenEnrichment_Settings
DragenRna_Settings
DragenSingleCellRna_Settings
DragenAmplicon_Settings
DragenGeoMxNGS_Settings
The run description can contain 255 alphanumeric characters, spaces, dashes, and underscores.
String with ASCII characters except for * and the control characters CR and LF. Enter NextSeq 1000 or NextSeq 2000.
RunName
No
The run name can contain 255 alphanumeric characters, spaces, dashes, and underscores.
String with ASCII characters except for * and the control characters CR and LF. Enter NextSeq 1000 or NextSeq 2000.
Read2Cycles
Yes
Number of cycles for Read 2. Required only when running a paired-end sequencing run.
Must be an integer ≥ 0.
Warning if values in range [1–25] inclusive.
If OverrideCycles is present in the BCL_Settings section, must be consistent with the sum of the Read2 section of OverrideCycles.
CustomRead2Primer
No¹
Indicates if a Custom Read 2 primer is used for the run.
Values true and false allowed. Value true only allowed if Read2Cycles is specified.
LibraryPrepKits
No
Identifies the library prep kit used for the run.
String with ASCII characters except for * and the control characters CR and LF.
Only one library prep kit is allowed.
In the NextSeq 1000/2000 Control Software v1.3 or later, the required custom recipe is automatically selected if one of the following kits is specified as the library prep kit.
BarcodeMismatchesIndex1
Optional
Specifies barcode mismatch tolerance for Index 1. Possible values are 0, 1, or 2, or na. The default value is 1. Only allowed if Index is specified in RunInfo.xml file and in the Reads section of the Sample Sheet.
A value of na must be used if:
BarcodeMismatchesIndex1 exists in the Data section of the Sample Sheet, and
OverrideCycles exist in the data section, and
BarcodeMismatchesIndex2
Optional
See description of BarcodeMismatchesIndex1, applied to BarcodeMismatchesIndex2.
OverrideCycles
Optional
Specifies the sequencing and indexing cycles to be used when processing the sequencing data. Must adhere to the following requirements: • Must be same number of fields (delimited by semicolon) as sequencing and indexing reads specified in RunInfo.xml and in the Reads section of the Sample Sheet. • Indexing reads are specified with I, sequencing reads are specified with Y, UMI cycles are specified with U, and trimmed reads are specified with N. • The number of cycles specified for each read must equal the number of cycles specified for that read in the RunInfo.xml file and in the Reads section of the Sample Sheet. • Only one Y or I sequence can be specified per read.
'I' cycles can only be specified for index reads
'Y' cycles can only be specified for genomic reads
FastqCompressionFormat
Optional
The compression format for the FASTQ output files. Allowed values are gzip or dragen.
NoLaneSplitting
Optional
If set to true, output all lanes of a flow cell to the same FASTQ files consecutively.
BCLConvert_Data
Sample_ID
Required
ID for the sample with the following requirements: • Must be alphanumeric string with _ or - and no spaces. • Case sensitive (i.e., a Sample Sheet with the samples MySample and mysample is not allowed)
The same Sample_ID may exist on more than one row of the Sample Sheet (e.g., one sample spanning more than one lane)
Undetermined is not allowed as a Sample_ID
Index
Optional
The Index 1 (i7) index sequence. Format is a sequence using ACTG. Required if index cycles are specified for the sample in the OverrideCycles.
Must adhere to the following requirements:
Can only contain A, C, G, or T
Length of string must match number of first index cycles in RunInfo.xml or the number specified in OverrideCycles
A value of na must be used if:
Index2
Optional
See description of Index, applied to Index2.
BCLConvert_Settings
SoftwareVersion
Required
The version of the software to be used to perform BCL conversion on the Sample_ID's that only exist in the BCL Convert Data section of the sample sheet., The version is specified using all three integers included in the DRAGEN version name. For example, 1.0.0.
AdapterRead1
Optional
The sequence of the Read 1 adapter to be masked or trimmed. To trim multiple adapters, separate the sequences with a plus sign (+) indicating independent adapters that must be independently assessed for masking or trimming for each read. Characters must be A, C, G, or T.
A value of na (case insensitive) must be used if:
the AdapterRead1 field is placed in the Data section of the Sample Sheet, and
no adapter trimming is desired for the sample
AdapterRead2
Optional
See description of AdapterRead1, applied to AdapterRead2.
BCLConvert_Data
BCLConvert_Settings
SoftwareVersion
Required
The version of the software to be used to perform BCL conversion on the Sample_ID's that only exist in the BCL Convert Data section of the sample sheet., The version is specified using all three integers included in the DRAGEN version name. For example, 1.0.0.
AdapterRead1
Optional
The sequence of the Read 1 adapter to be masked or trimmed. To trim multiple adapters, separate the sequences with a plus sign (+) indicating independent adapters that must be independently assessed for masking or trimming for each read. Characters must be A, C, G, or T.
A value of na (case insensitive) must be used if:
the AdapterRead1 field is placed in the Data section of the Sample Sheet, and
no adapter trimming is desired for the sample
AdapterRead2
Optional
See description of AdapterRead1, applied to AdapterRead2.
BCLConvert_Data
BCLConvert_Settings
SoftwareVersion
Required
The version of the software to be used to perform BCL conversion on the Sample_ID's that only exist in the BCL Convert Data section of the sample sheet., The version is specified using all three integers included in the DRAGEN version name. For example, 1.0.0.
AdapterRead1
Optional
The sequence of the Read 1 adapter to be masked or trimmed. To trim multiple adapters, separate the sequences with a plus sign (+) indicating independent adapters that must be independently assessed for masking or trimming for each read. Characters must be A, C, G, or T.
A value of na (case insensitive) must be used if:
the AdapterRead1 field is placed in the Data section of the Sample Sheet, and
no adapter trimming is desired for the sample
AdapterRead2
Optional
See description of AdapterRead1, applied to AdapterRead2.
BCLConvert_Data
BCLConvert_Settings
SoftwareVersion
Required
The version of the software to be used to perform BCL conversion on the Sample_ID's that only exist in the BCL Convert Data section of the sample sheet., The version is specified using all three integers included in the DRAGEN version name. For example, 1.0.0.
AdapterRead1
Optional
The sequence of the Read 1 adapter to be masked or trimmed. To trim multiple adapters, separate the sequences with a plus sign (+) indicating independent adapters that must be independently assessed for masking or trimming for each read. Characters must be A, C, G, or T.
A value of na (case insensitive) must be used if:
the AdapterRead1 field is placed in the Data section of the Sample Sheet, and
no adapter trimming is desired for the sample
AdapterRead2
Optional
See description of AdapterRead1, applied to AdapterRead2.
BCLConvert_Data
BCLConvert_Settings
SoftwareVersion
Required
The version of the software to be used to perform BCL conversion on the Sample_ID's that only exist in the BCL Convert Data section of the sample sheet., The version is specified using all three integers included in the DRAGEN version name. For example, 1.0.0.
AdapterRead1
Optional
The sequence of the Read 1 adapter to be masked or trimmed. To trim multiple adapters, separate the sequences with a plus sign (+) indicating independent adapters that must be independently assessed for masking or trimming for each read. Characters must be A, C, G, or T.
A value of na (case insensitive) must be used if:
the AdapterRead1 field is placed in the Data section of the Sample Sheet, and
no adapter trimming is desired for the sample
AdapterRead2
Optional
See description of AdapterRead1, applied to AdapterRead2.
BCLConvert_Data
KeepFastQ
Optional
Select whether FASTQs are saved (true) or discarded (false).
DragenGermline_Data
Sample_ID
Required
See description in the BCL Convert section
DragenGermline_Settings
SoftwareVersion
Required
The version of the DRAGEN software~~,~~ used to process the DragenGermline pipeline, including conversion to FASTQ. Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
ReferenceGenomeDir
Required
Genome name consisting of alphanumeric string with "_" or "-" or ".".
MapAlignOutFormat
Optional
Formatting of the output files. Accepted values are bam, cram, or none. Default is bam.
DragenGermline_Data
BCLConvert_Settings
SoftwareVersion
Required
The version of the software to be used to perform BCL conversion on the Sample_ID's that only exist in the BCL Convert Data section of the sample sheet., The version is specified using all three integers included in the DRAGEN version name. For example, 1.0.0.
AdapterRead1
Optional
The sequence of the Read 1 adapter to be masked or trimmed. To trim multiple adapters, separate the sequences with a plus sign (+) indicating independent adapters that must be independently assessed for masking or trimming for each read. Characters must be A, C, G, or T.
A value of na (case insensitive) must be used if:
the AdapterRead1 field is placed in the Data section of the Sample Sheet, and
no adapter trimming is desired for the sample
AdapterRead2
Optional
See description of AdapterRead1, applied to AdapterRead2.
BCLConvert_Data
DragenGermline_Settings
SoftwareVersion
Required
The version of the DRAGEN software~~,~~ used to process the DragenGermline pipeline, including conversion to FASTQ, Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
ReferenceGenomeDir
Required
Genome name consisting of alphanumeric string with "_" or "-" or ".".
MapAlignOutFormat
Optional
Formatting of the output files. Accepted values are bam, cram, or none. Default is bam.
DragenGermline_Data
DragenGermline_Settings
SoftwareVersion
Required
The version of the DRAGEN software~~,~~ used to process the DragenGermline pipeline, including conversion to FASTQ, Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
ReferenceGenomeDir
Required
Genome name consisting of alphanumeric string with "_" or "-" or ".".
MapAlignOutFormat
Optional
Formatting of the output files. Accepted values are bam, cram, or none. Default is bam.
DragenGermline_Data
DragenGermline_Settings
SoftwareVersion
Required
The version of the DRAGEN software~~,~~ used to process the DragenGermline pipeline, including conversion to FASTQ, Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
ReferenceGenomeDir
Required
Genome name consisting of alphanumeric string with "_" or "-" or ".".
MapAlignOutFormat
Optional
Formatting of the output files. Accepted values are bam, cram, or none. Default is bam.
DragenGermline_Data
Bedfile
Required
BED file to be used for analysis in text (*.txt) or gzip (*.gz) format. Alphanumeric string with _ or - or . with no spaces allowed.
KeepFastQ
Optional
Select whether FASTQs are saved (true) or discarded (false).
GermlineOrSomatic
Required
Indicator of pipeline type. Accepted values are germline or somatic.
AuxNoiseBaselineFile
Optional
Alphanumeric string with "_" or "-" or ".". and no spaces in text (*.txt) or gzip (*.gz) format. Applicable only if GermlineOrSomatic is Somatic.
AuxCnvPanelOfNormalsFile
Optional
Alphanumeric string with "_" or "-" or "." and no spaces in text (*.txt) or gzip (*.gz) format.
DragenEnrichment_Data
Sample_ID
Required
See description in the BCL Convert section
DragenEnrichment_Settings
SoftwareVersion
Required
The version of the DRAGEN software used to process the DragenEnrichment pipeline, including conversion to FASTQ. Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
MapAlignOutFormat
Optional
Formatting of the output files. Accepted values are bam, cram, or none. Default is bam.
ReferenceGenomeDir
Required
Genome name consisting of alphanumeric string with "_" or "-" or ".".
DragenEnrichment_Data
DragenEnrichment_Settings
SoftwareVersion
Required
The version of the DRAGEN software used to process the DragenEnrichment pipeline, including conversion to FASTQ. Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
MapAlignOutFormat
Optional
Formatting of the output files. Accepted values are bam, cram, or none. Default is bam.
ReferenceGenomeDir
Required
Genome name consisting of alphanumeric string with "_" or "-" or ".".
DragenEnrichment_Data
DragenEnrichment_Settings
SoftwareVersion
Required
The version of the DRAGEN software used to process the DragenEnrichment pipeline, including conversion to FASTQ. Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
MapAlignOutFormat
Optional
Formatting of the output files. Accepted values are bam, cram, or none. Default is bam.
ReferenceGenomeDir
Required
Genome name consisting of alphanumeric string with "_" or "-" or ".".
DragenEnrichment_Data
DragenEnrichment_Settings
SoftwareVersion
Required
The version of the DRAGEN software used to process the DragenEnrichment pipeline, including conversion to FASTQ. Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
MapAlignOutFormat
Optional
Formatting of the output files. Accepted values are bam, cram, or none. Default is bam.
ReferenceGenomeDir
Required
Genome name consisting of alphanumeric string with "_" or "-" or ".".
DragenEnrichment_Data
RnaGeneAnnotationFile
Optional
Genotype reference file. Alphanumeric string with "_" or "-" or "." with no spaces allowed. If DifferentialExpressionEnable is True., the GTF file must be provided by the user or included with the reference genome.
KeepFastQ
Optional
Select whether FASTQs are saved (true) or discarded (false).
DifferentialExpressionEnable
Optional
Accepted values are true or false.
DragenRna_Data
Sample_ID
Required
See description in the BCL Convert section
Comparison1
Optional
Accepted values are control, comparison, or na. Use only if DifferentialExpressionEnable is true. Must have at least 2 control and 2 comparison samples. May have a max of 15 control or comparison samples.
Comparison2
Optional
See Comparison1 description.
Comparison3
Optional
DragenRna_Settings
SoftwareVersion
Required
The version of the DRAGEN software used to process the DragenRNA pipeline, including conversion to FASTQ. Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
MapAlignOutFormat
Optional
Formatting of the output files. Accepted values are bam, cram, or none. Default is bam.
ReferenceGenomeDir
Required
Genome name consisting of alphanumeric string with "_" or "-" or ".".
DragenRna_Data
DragenRna_Settings
SoftwareVersion
Required
The version of the DRAGEN software used to process the DragenRNA pipeline, including conversion to FASTQ. Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
MapAlignOutFormat
Optional
Formatting of the output files. Accepted values are bam, cram, or none. Default is bam.
ReferenceGenomeDir
Required
Genome name consisting of alphanumeric string with "_" or "-" or ".".
DragenRna_Data
DragenRna_Settings
SoftwareVersion
Required
The version of the DRAGEN software used to process the DragenRNA pipeline, including conversion to FASTQ. Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
MapAlignOutFormat
Optional
Formatting of the output files. Accepted values are bam, cram, or none. Default is bam.
ReferenceGenomeDir
Required
Genome name consisting of alphanumeric string with "_" or "-" or ".".
DragenRna_Data
DragenRna_Settings
SoftwareVersion
Required
The version of the DRAGEN software used to process the DragenRNA pipeline, including conversion to FASTQ. Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
MapAlignOutFormat
Optional
Formatting of the output files. Accepted values are bam, cram, or none. Default is bam.
ReferenceGenomeDir
Required
Genome name consisting of alphanumeric string with "_" or "-" or ".".
DragenRna_Data
RnaGeneAnnotationFile
Optional
Genotype reference file. Alphanumeric string with "_" or "-" or "." with no spaces allowed. If DifferentialExpressionEnable is True., the GTF file must be provided by the user or included with the reference genome.
KeepFastQ
Optional
Select whether FASTQs are saved (true) or discarded (false).
BarcodePosition
Required
Text string indicating the location of the bases corresponding to the Barcode within the specified BarcodeRead. Base positions are indexed starting at position zero in the Read. Examples: 0_15, or 0_8+21_29+43_51 if the barcodes are not contiguous.
UmiPosition
Required
Text string indicating the location of the bases corresponding to the UMI within the specified BarcodeRead. Base positions are indexed starting at position zero in the Read. Example: 16_27
BarcodeRead
Required
Text string indicating the location within the sequencing run of the Barcode Read, which contains both the Barcode and the Umi. Readn, where n is from the set { 1, 2 }
BarcodeSequenceWhiteList
Required
List of expected barcode sequences
BarcodeSequenceList
Required
List of expected barcode sequences
RnaLibraryType
Required
Valid values are StrandedForward, StrandedReverse, Unstranded
DragenSingleCellRna_Data
Sample_ID
Required
See description in the BCL Convert section
DragenSingleCellRna_Settings
SoftwareVersion
Required
The version of the DRAGEN software used to process the DragenSingleCellRNA pipeline, including conversion to FASTQ. Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
MapAlignOutFormat
Optional
Formatting of the output files. Accepted values are bam, cram, or none. Default is bam.
ReferenceGenomeDir
Required
Genome name consisting of alphanumeric string with "_" or "-" or ".".
DragenSingleCellRna_Data
DragenSingleCellRna_Settings
SoftwareVersion
Required
The version of the DRAGEN software used to process the DragenSingleCellRNA pipeline, including conversion to FASTQ. Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
MapAlignOutFormat
Optional
Formatting of the output files. Accepted values are bam, cram, or none. Default is bam.
ReferenceGenomeDir
Required
Genome name consisting of alphanumeric string with "_" or "-" or ".".
DragenSingleCellRna_Data
DragenSingleCellRna_Settings
SoftwareVersion
Required
The version of the DRAGEN software used to process the DragenSingleCellRNA pipeline, including conversion to FASTQ. Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
MapAlignOutFormat
Optional
Formatting of the output files. Accepted values are bam, cram, or none. Default is bam.
ReferenceGenomeDir
Required
Genome name consisting of alphanumeric string with "_" or "-" or ".".
DragenSingleCellRna_Data
DragenSingleCellRna_Settings
SoftwareVersion
Required
The version of the DRAGEN software used to process the DragenSingleCellRNA pipeline, including conversion to FASTQ. Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
MapAlignOutFormat
Optional
Formatting of the output files. Accepted values are bam, cram, or none. Default is bam.
ReferenceGenomeDir
Required
Genome name consisting of alphanumeric string with "_" or "-" or ".".
DragenSingleCellRna_Data
KeepFastQ
Optional
Select whether FASTQs are saved (true) or discarded (false).
DnaBedFile
Required
BED file to be used for analysis in text (*.txt) or gzip (*.gz) format. Alphanumeric string with _ or - or . with no spaces allowed.
DnaGermlineOrSomatic
Required
Indicator of "germline" or "somatic" type for DNA samples
DragenSingleCellRna_Data
Sample_ID
Required
See description in the BCL Convert section
DnaOrRna
Required
Indicator of a DNA or RNA sample. Valid values are dna and rna.
DragenAmplicon_Settings
SoftwareVersion
Required
The version of the DRAGEN software used to process the DragenAmplicon pipeline, including conversion to FASTQ. Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
MapAlignOutFormat
Optional
Formatting of the output files. Accepted values are bam, cram, or none. Default is bam.
ReferenceGenomeDir
Required
Genome name consisting of alphanumeric string with "_" or "-" or ".".
DragenSingleCellRna_Data
DragenAmplicon_Settings
SoftwareVersion
Required
The version of the DRAGEN software used to process the DragenAmplicon pipeline, including conversion to FASTQ. Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
MapAlignOutFormat
Optional
Formatting of the output files. Accepted values are bam, cram, or none. Default is bam.
ReferenceGenomeDir
Required
Genome name consisting of alphanumeric string with "_" or "-" or ".".
DragenSingleCellRna_Data
DragenAmplicon_Settings
SoftwareVersion
Required
The version of the DRAGEN software used to process the DragenAmplicon pipeline, including conversion to FASTQ. Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
MapAlignOutFormat
Optional
Formatting of the output files. Accepted values are bam, cram, or none. Default is bam.
ReferenceGenomeDir
Required
Genome name consisting of alphanumeric string with "_" or "-" or ".".
DragenSingleCellRna_Data
KeepFastq
Optional
Select whether FASTQs are saved (true) or discarded (false).
DragenGeoMxNGS_Data
Sample_ID
Required
See description in the BCL Convert section
DragenGeoMxNGS_Settings
SoftwareVersion
Required
The version of the DRAGEN software~~,~~ used to process the DragenGeoMxNGS pipeline, including conversion to FASTQ. Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
GeoMxConfigFile
Required
GeoMx configuration file
BarcodeSequenceList
Required
List of expected barcode sequences
DragenGeoMxNGS_Data
Custom_*
No
Custom field used to capture run metadata.
String with ASCII characters except for * and the control characters CR and LF.
FileFormatVersion
Yes
Used to identify the sample sheet as a v2 sample sheet. This field must always exist in the header section with a value of 2.
Must always be 2.
InstrumentPlatform
No
Identifies the instrument platform to be used for the run.
For example, enter NextSeq1k2k for NextSeq 1000/2000 Series.
String with ASCII characters except for * and the control characters CR and LF.
InstrumentType
No
Identifies the instrument to be used for the run.
For example: if using NextSeq 2000, populate the field with NextSeq2000.
String with ASCII characters except for * and the control characters CR and LF.
RunDescription
Index1Cycles
No
Number of cycles in Index Read 1. Required if more than one sample is present in sample sheet.
Must be an integer ≥ 0.
Depending on your sequencing system and reagent kit, there can be limitations on the number of cycles in the Index Read.
Warning if values in range [1–5] inclusive.
If there is more than 1 sample per lane, must be > 0.
If OverrideCycles is present in the BCL_Settings section, must be consistent with the sum of the Index1 section of OverrideCycles.
Index2Cycles
No
Number of cycles in Index Read 2. Required if using dual indexes for demultiplexing.
Must be an integer ≥ 0.
Depending on your sequencing system and reagent kit, there can be limitations on the number of cycles in the Index Read.
Warning if values in range [1–5] inclusive.
If OverrideCycles is present in the BCL_Settings section, must be consistent with the sum of the Index1 section of OverrideCycles.
Read1Cycles
Yes
Number of cycles for Read 1.
CustomIndex1Primer
No¹
Indicates if a Custom Index 1 primer is used for the run.
Values true and false allowed. Value true only allowed if Index1Cycles is specified.
CustomIndex2Primer
No¹
Indicates if a Custom Index 2 primer is used for the run.
Values true and false allowed. Value true only allowed if Index2Cycles is specified.
CustomRead1Primer
No¹
Indicates if a Custom Read 1 primer is used for the run.
SoftwareVersion
Required
The version of the software to be used to perform BCL conversion on the Sample_ID's that only exist in the BCL Convert Data section of the sample sheet., The version is specified using all three integers included in the DRAGEN version name. For example, 1.0.0.
AdapterRead1
Optional
The sequence of the Read 1 adapter to be masked or trimmed. To trim multiple adapters, separate the sequences with a plus sign (+) indicating independent adapters that must be independently assessed for masking or trimming for each read. Characters must be A, C, G, or T.
A value of na (case insensitive) must be used if:
the AdapterRead1 field is placed in the Data section of the Sample Sheet, and
no adapter trimming is desired for the sample
AdapterRead2
Optional
See description of AdapterRead1, applied to AdapterRead2.
SoftwareVersion
Required
The version of the DRAGEN software used to process the DragenGermline pipeline, including conversion to FASTQ. Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
ReferenceGenomeDir
Required
Genome name consisting of alphanumeric string with "_" or "-" or ".".
MapAlignOutFormat
Optional
Formatting of the output files. Accepted values are bam, cram, or none. Default is bam.
SoftwareVersion
Required
The version of the DRAGEN software used to process the DragenEnrichment pipeline, including conversion to FASTQ. Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
MapAlignOutFormat
Optional
Formatting of the output files. Accepted values are bam, cram, or none. Default is bam.
ReferenceGenomeDir
Required
Genome name consisting of alphanumeric string with "_" or "-" or ".".
SoftwareVersion
Required
The version of the DRAGEN software used to process the DragenRNA pipeline, including conversion to FASTQ. Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
MapAlignOutFormat
Optional
Formatting of the output files. Accepted values are bam, cram, or none. Default is bam.
ReferenceGenomeDir
Required
Genome name consisting of alphanumeric string with "_" or "-" or ".".
SoftwareVersion
Required
The version of the DRAGEN software used to process the DragenSingleCellRNA pipeline, including conversion to FASTQ. Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
MapAlignOutFormat
Optional
Formatting of the output files. Accepted values are bam, cram, or none. Default is bam.
ReferenceGenomeDir
Required
Genome name consisting of alphanumeric string with "_" or "-" or ".".
SoftwareVersion
Required
The version of the DRAGEN software used to process the DragenAmplicon pipeline, including conversion to FASTQ. Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
MapAlignOutFormat
Optional
Formatting of the output files. Accepted values are bam, cram, or none. Default is bam.
ReferenceGenomeDir
Required
Genome name consisting of alphanumeric string with "_" or "-" or ".".
SoftwareVersion
Required
The version of the DRAGEN software~~,~~ used to process the DragenGeoMxNGS pipeline, including conversion to FASTQ. Specified using all three integers included in the DRAGEN version name. For example 4.1.5.
GeoMxConfigFile
Required
GeoMx configuration file
BarcodeSequenceList
Required
List of expected barcode sequences
No
Must be an integer > 0.
Warning if less than 26.
If OverrideCycles is present in the BCL_Settings section, must be consistent with the sum of the Read1 section of OverrideCycles.
Values true and false allowed. Value true only allowed if Read1Cycles is specified.
Comparison4
Optional
See Comparison1 description.
Comparison5
Optional
See Comparison1 description.
Comparison4
Optional
See Comparison1 description.
Comparison5
Optional
See Comparison1 description.
Comparison4
Optional
See Comparison1 description.
Comparison5
Optional
See Comparison1 description.
Illumina Stranded Total RNA Prep with Ribo-Zero Plus kit
Illumina Stranded mRNA Prep kit
Enter one of the following values.
For Illumina Stranded Total RNA Prep with Ribo-Zero Plus kit, enter ILMNStrandedTotalRNA.
For Illumina Stranded mRNA Prep kit, enter ILMNStrandedmRNA.
The total number of cycles used for demultiplexing by both indexes together cannot exceed 27. Note that this includes all cycles between the first "I" cycle used by any sample and the last "I" cycle used by any sample within each index.
"I5N3;I5N3" : counts as 10 bases toward the limit of 27
"I4N1I3;I5N3": counts as (8+5) 13 bases toward the limit of 27
The following are examples of OverrideCycles input: U8Y143;I8;I8;U8Y143 N10Y66;I6;N10Y66
For a sample sheet containing two samples having the following OverrideCycles
Y151;I8N2;N10;Y151
Y151;N2I8;I8N2;Y151
the number of cycles used for demultiplexing sums to 18.
For sequencers with RunInfo.xml having "IsReverseComplement" set to "Y" for Index2, then Index2 is processed in reverse complement orientation. Any trimming should be applied to the end of the sequence as it transitions to the adapter, which means the "N" cycles specified in OverrideCycles must be in the beginning of the index read.
Example of 8bp i5 index with the last two (adapter) bases to be trimmed:
Forward i5: XXATCGCGGT
ReverseComp i5: ACCGCGATXX (this is the direction of sequencing)
OverrideCycles for Index2: N2I8
where XX is part of the adapter sequence.
Although Index2 is processed on the sequencer in reverse complement format, Index2 and OverrideCycles are entered in the Sample Sheet in forward, non-complemented format for user convenience (see diagram below).
Note that NovaSeq X has a RunInfo.xml with "IsReverseComplement" set to "Y" for Index2.
No indexes are specified in OverrideCycles for a sample
BarcodeMismatchesIndex1
Optional
Specifies barcode mismatch tolerance for Index 1. Possible values are 0, 1, or 2, or na. The default value is 1. Only allowed if Index is specified in RunInfo.xml file and in the Reads section of the Sample Sheet.
A value of na must be used if:
BarcodeMismatchesIndex1 exists in the Data section of the Sample Sheet, and
OverrideCycles exist in the data section, and
Index 1 is masked out for the sample in the OverrideCycles
BarcodeMismatchesIndex2
Optional
See description of BarcodeMismatchesIndex1, applied to BarcodeMismatchesIndex2.
OverrideCycles
Optional
Specifies the sequencing and indexing cycles to be used when processing the sequencing data. Must adhere to the following requirements:
• Must be same number of fields (delimited by
semicolon) as sequencing and indexing reads specified in RunInfo.xml or and in the Reads section of the Sample Sheet.
• Indexing reads are specified with I, sequencing reads are specified with Y, UMI cycles are specified with U, and trimmed
reads are specified with N.
• The number of cycles specified for each read must equal the number of cycles specified for that read in the RunInfo.xml file and in the Reads section of the Sample Sheet.
• Only one Y or I sequence can be specified per read.
'I' cycles can only be specified for index reads
'Y' cycles can only be specified for genomic reads
The total number of cycles used for demultiplexing by both indexes together cannot exceed 27. Note that this includes all cycles between the first "I" cycle used by any sample and the last "I" cycle used by any sample within each index.
"I5N3;I5N3" : counts as 10 bases toward the limit of 27
"I4N1I3;I5N3": counts as (8+5) 13 bases toward the limit of 27
The following are examples of OverrideCycles input: U8Y143;I8;I8;U8Y143 N10Y66;I6;N10Y66
For a sample sheet containing two samples having the following OverrideCycles
Y151; I8N2; N10; Y151
Y151; N2I8; I8N2; Y151
the number of cycles used for demultiplexing sums to 18.
For sequencers with RunInfo.xml having "IsReverseComplement" set to "Y" for Index2, then Index2 is processed in reverse complement orientation. Any trimming should be applied to the end of the sequence as it transitions to the adapter, which means the "N" cycles specified in OverrideCycles must be in the beginning of the index read.
Example of 8bp i5 index with the last two (adapter) bases to be trimmed:
Forward i5: XXATCGCGGT
ReverseComp i5: ACCGCGATXX (this is the direction of sequencing)
OverrideCycles for Index2: N2I8
where XX is part of the adapter sequence.
Although Index2 is processed on the sequencer in reverse complement format, Index2 and OverrideCycles are entered in the Sample Sheet in forward, non-complemented format for user convenience (see diagram below).
Note that NovaSeq X has a RunInfo.xml with "IsReverseComplement" set to "Y" for Index2,
FastqCompressionFormat
Optional
The compression format for the FASTQ output files. Allowed values are gzip or dragen.
Sample_ID
Required
ID for the sample with the following requirements: • Must be alphanumeric string with _ or - and no spaces. • Case sensitive (i.e., a Sample Sheet with the samples MySample and mysample is not allowed)
The same Sample_ID may exist on more than one row of the Sample Sheet (e.g., one sample spanning more than one lane)
Undetermined is not allowed as a Sample_ID
Index
Optional
The Index 1 (i7) index sequence. Format is a sequence using ACTG. Required if index cycles are specified for the sample in the OverrideCycles.
Must adhere to the following requirements:
Can only contain A, C, G, or T
Length of string must match number of first index cycles in RunInfo.xml or the number specified in OverrideCycles
A value of na must be used if:
No indexes are specified in OverrideCycles for a sample
Index2
Optional
See description of Index, applied to Index2.
BarcodeMismatchesIndex1
Optional
Specifies barcode mismatch tolerance for Index 1. Possible values are 0, 1, or 2, or na. The default value is 1. Only allowed if Index is specified in RunInfo.xml file and in the Reads section of the Sample Sheet.
A value of na must be used if:
BarcodeMismatchesIndex1 exists in the Data section of the Sample Sheet, and
OverrideCycles exist in the data section, and
Index 1 is masked out for the sample in the OverrideCycles
BarcodeMismatchesIndex2
Optional
See description of BarcodeMismatchesIndex1, applied to BarcodeMismatchesIndex2.
OverrideCycles
Optional
Specifies the sequencing and indexing cycles to be used when processing the sequencing data. Must adhere to the following requirements:
• Must be same number of fields (delimited by
semicolon) as sequencing and indexing reads specified in RunInfo.xml or and in the Reads section of the Sample Sheet.
• Indexing reads are specified with I, sequencing reads are specified with Y, UMI cycles are specified with U, and trimmed
reads are specified with N.
• The number of cycles specified for each read must equal the number of cycles specified for that read in the RunInfo.xml file and in the Reads section of the Sample Sheet.
• Only one Y or I sequence can be specified per read.
'I' cycles can only be specified for index reads
'Y' cycles can only be specified for genomic reads
The total number of cycles used for demultiplexing by both indexes together cannot exceed 27. Note that this includes all cycles between the first "I" cycle used by any sample and the last "I" cycle used by any sample within each index.
"I5N3;I5N3" : counts as 10 bases toward the limit of 27
"I4N1I3;I5N3": counts as (8+5) 13 bases toward the limit of 27
The following are examples of OverrideCycles input: U8Y143;I8;I8;U8Y143 N10Y66;I6;N10Y66
For a sample sheet containing two samples having the following OverrideCycles
Y151; I8N2; N10; Y151
Y151; N2I8; I8N2; Y151
the number of cycles used for demultiplexing sums to 18.
For sequencers with RunInfo.xml having "IsReverseComplement" set to "Y" for Index2, then Index2 is processed in reverse complement orientation. Any trimming should be applied to the end of the sequence as it transitions to the adapter, which means the "N" cycles specified in OverrideCycles must be in the beginning of the index read.
Example of 8bp i5 index with the last two (adapter) bases to be trimmed:
Forward i5: XXATCGCGGT
ReverseComp i5: ACCGCGATXX (this is the direction of sequencing)
OverrideCycles for Index2: N2I8
where XX is part of the adapter sequence.
Although Index2 is processed on the sequencer in reverse complement format, Index2 and OverrideCycles are entered in the Sample Sheet in forward, non-complemented format for user convenience (see diagram below).
Note that NovaSeq X has a RunInfo.xml with "IsReverseComplement" set to "Y" for Index2,
FastqCompressionFormat
Optional
The compression format for the FASTQ output files. Allowed values are gzip or dragen.
Sample_ID
Required
ID for the sample with the following requirements: • Must be alphanumeric string with _ or - and no spaces. • Case sensitive (i.e., a Sample Sheet with the samples MySample and mysample is not allowed)
The same Sample_ID may exist on more than one row of the Sample Sheet (e.g., one sample spanning more than one lane)
Undetermined is not allowed as a Sample_ID
Index
Optional
The Index 1 (i7) index sequence. Format is a sequence using ACTG. Required if index cycles are specified for the sample in the OverrideCycles.
Must adhere to the following requirements:
Can only contain A, C, G, or T
Length of string must match number of first index cycles in RunInfo.xml or the number specified in OverrideCycles
A value of na must be used if:
No indexes are specified in OverrideCycles for a sample
Index2
Optional
See description of Index, applied to Index2.
BarcodeMismatchesIndex1
Optional
Specifies barcode mismatch tolerance for Index 1. Possible values are 0, 1, or 2, or na. The default value is 1. Only allowed if Index is specified in RunInfo.xml file and in the Reads section of the Sample Sheet.
A value of na must be used if:
BarcodeMismatchesIndex1 exists in the Data section of the Sample Sheet, and
OverrideCycles exist in the data section, and
Index 1 is masked out for the sample in the OverrideCycles
BarcodeMismatchesIndex2
Optional
See description of BarcodeMismatchesIndex1, applied to BarcodeMismatchesIndex2.
OverrideCycles
Optional
Specifies the sequencing and indexing cycles to be used when processing the sequencing data. Must adhere to the following requirements:
• Must be same number of fields (delimited by
semicolon) as sequencing and indexing reads specified in RunInfo.xml or and in the Reads section of the Sample Sheet.
• Indexing reads are specified with I, sequencing reads are specified with Y, UMI cycles are specified with U, and trimmed
reads are specified with N.
• The number of cycles specified for each read must equal the number of cycles specified for that read in the RunInfo.xml file and in the Reads section of the Sample Sheet.
• Only one Y or I sequence can be specified per read.
'I' cycles can only be specified for index reads
'Y' cycles can only be specified for genomic reads
The total number of cycles used for demultiplexing by both indexes together cannot exceed 27. Note that this includes all cycles between the first "I" cycle used by any sample and the last "I" cycle used by any sample within each index.
"I5N3;I5N3" : counts as 10 bases toward the limit of 27
"I4N1I3;I5N3": counts as (8+5) 13 bases toward the limit of 27
The following are examples of OverrideCycles input: U8Y143;I8;I8;U8Y143 N10Y66;I6;N10Y66
For a sample sheet containing two samples having the following OverrideCycles
Y151; I8N2; N10; Y151
Y151; N2I8; I8N2; Y151
the number of cycles used for demultiplexing sums to 18.
For sequencers with RunInfo.xml having "IsReverseComplement" set to "Y" for Index2, then Index2 is processed in reverse complement orientation. Any trimming should be applied to the end of the sequence as it transitions to the adapter, which means the "N" cycles specified in OverrideCycles must be in the beginning of the index read.
Example of 8bp i5 index with the last two (adapter) bases to be trimmed:
Forward i5: XXATCGCGGT
ReverseComp i5: ACCGCGATXX (this is the direction of sequencing)
OverrideCycles for Index2: N2I8
where XX is part of the adapter sequence.
Although Index2 is processed on the sequencer in reverse complement format, Index2 and OverrideCycles are entered in the Sample Sheet in forward, non-complemented format for user convenience (see diagram below).
Note that NovaSeq X has a RunInfo.xml with "IsReverseComplement" set to "Y" for Index2,
FastqCompressionFormat
Optional
The compression format for the FASTQ output files. Allowed values are gzip or dragen.
Sample_ID
Required
ID for the sample with the following requirements: • Must be alphanumeric string with _ or - and no spaces. • Case sensitive (i.e., a Sample Sheet with the samples MySample and mysample is not allowed)
The same Sample_ID may exist on more than one row of the Sample Sheet (e.g., one sample spanning more than one lane)
Undetermined is not allowed as a Sample_ID
Index
Optional
The Index 1 (i7) index sequence. Format is a sequence using ACTG. Required if index cycles are specified for the sample in the OverrideCycles.
Must adhere to the following requirements:
Can only contain A, C, G, or T
Length of string must match number of first index cycles in RunInfo.xml or the number specified in OverrideCycles
A value of na must be used if:
No indexes are specified in OverrideCycles for a sample
Index2
Optional
See description of Index, applied to Index2.
BarcodeMismatchesIndex1
Optional
Specifies barcode mismatch tolerance for Index 1. Possible values are 0, 1, or 2, or na. The default value is 1. Only allowed if Index is specified in RunInfo.xml file and in the Reads section of the Sample Sheet.
A value of na must be used if:
BarcodeMismatchesIndex1 exists in the Data section of the Sample Sheet, and
OverrideCycles exist in the data section, and
Index 1 is masked out for the sample in the OverrideCycles
BarcodeMismatchesIndex2
Optional
See description of BarcodeMismatchesIndex1, applied to BarcodeMismatchesIndex2.
OverrideCycles
Optional
Specifies the sequencing and indexing cycles to be used when processing the sequencing data. Must adhere to the following requirements:
• Must be same number of fields (delimited by
semicolon) as sequencing and indexing reads specified in RunInfo.xml or and in the Reads section of the Sample Sheet.
• Indexing reads are specified with I, sequencing reads are specified with Y, UMI cycles are specified with U, and trimmed
reads are specified with N.
• The number of cycles specified for each read must equal the number of cycles specified for that read in the RunInfo.xml file and in the Reads section of the Sample Sheet.
• Only one Y or I sequence can be specified per read.
'I' cycles can only be specified for index reads
'Y' cycles can only be specified for genomic reads
The total number of cycles used for demultiplexing by both indexes together cannot exceed 27. Note that this includes all cycles between the first "I" cycle used by any sample and the last "I" cycle used by any sample within each index.
"I5N3;I5N3" : counts as 10 bases toward the limit of 27
"I4N1I3;I5N3": counts as (8+5) 13 bases toward the limit of 27
The following are examples of OverrideCycles input: U8Y143;I8;I8;U8Y143 N10Y66;I6;N10Y66
For a sample sheet containing two samples having the following OverrideCycles
Y151; I8N2; N10; Y151
Y151; N2I8; I8N2; Y151
the number of cycles used for demultiplexing sums to 18.
For sequencers with RunInfo.xml having "IsReverseComplement" set to "Y" for Index2, then Index2 is processed in reverse complement orientation. Any trimming should be applied to the end of the sequence as it transitions to the adapter, which means the "N" cycles specified in OverrideCycles must be in the beginning of the index read.
Example of 8bp i5 index with the last two (adapter) bases to be trimmed:
Forward i5: XXATCGCGGT
ReverseComp i5: ACCGCGATXX (this is the direction of sequencing)
OverrideCycles for Index2: N2I8
where XX is part of the adapter sequence.
Although Index2 is processed on the sequencer in reverse complement format, Index2 and OverrideCycles are entered in the Sample Sheet in forward, non-complemented format for user convenience (see diagram below).
Note that NovaSeq X has a RunInfo.xml with "IsReverseComplement" set to "Y" for Index2,
FastqCompressionFormat
Optional
The compression format for the FASTQ output files. Allowed values are gzip or dragen.
Sample_ID
Required
ID for the sample with the following requirements: • Must be alphanumeric string with _ or - and no spaces. • Case sensitive (i.e., a Sample Sheet with the samples MySample and mysample is not allowed)
The same Sample_ID may exist on more than one row of the Sample Sheet (e.g., one sample spanning more than one lane)
Undetermined is not allowed as a Sample_ID
Index
Optional
The Index 1 (i7) index sequence. Format is a sequence using ACTG. Required if index cycles are specified for the sample in the OverrideCycles.
Must adhere to the following requirements:
Can only contain A, C, G, or T
Length of string must match number of first index cycles in RunInfo.xml or the number specified in OverrideCycles
A value of na must be used if:
No indexes are specified in OverrideCycles for a sample
Index2
Optional
See description of Index, applied to Index2.
BarcodeMismatchesIndex1
Optional
Specifies barcode mismatch tolerance for Index 1. Possible values are 0, 1, or 2, or na. The default value is 1. Only allowed if Index is specified in RunInfo.xml file and in the Reads section of the Sample Sheet.
A value of na must be used if:
BarcodeMismatchesIndex1 exists in the Data section of the Sample Sheet, and
OverrideCycles exist in the data section, and
Index 1 is masked out for the sample in the OverrideCycles
BarcodeMismatchesIndex2
Optional
See description of BarcodeMismatchesIndex1, applied to BarcodeMismatchesIndex2.
OverrideCycles
Optional
Specifies the sequencing and indexing cycles to be used when processing the sequencing data. Must adhere to the following requirements:
• Must be same number of fields (delimited by
semicolon) as sequencing and indexing reads specified in RunInfo.xml or and in the Reads section of the Sample Sheet.
• Indexing reads are specified with I, sequencing reads are specified with Y, UMI cycles are specified with U, and trimmed
reads are specified with N.
• The number of cycles specified for each read must equal the number of cycles specified for that read in the RunInfo.xml file and in the Reads section of the Sample Sheet.
• Only one Y or I sequence can be specified per read.
'I' cycles can only be specified for index reads
'Y' cycles can only be specified for genomic reads
The total number of cycles used for demultiplexing by both indexes together cannot exceed 27. Note that this includes all cycles between the first "I" cycle used by any sample and the last "I" cycle used by any sample within each index.
"I5N3;I5N3" : counts as 10 bases toward the limit of 27
"I4N1I3;I5N3": counts as (8+5) 13 bases toward the limit of 27
The following are examples of OverrideCycles input: U8Y143;I8;I8;U8Y143 N10Y66;I6;N10Y66
For a sample sheet containing two samples having the following OverrideCycles
Y151; I8N2; N10; Y151
Y151; N2I8; I8N2; Y151
the number of cycles used for demultiplexing sums to 18.
For sequencers with RunInfo.xml having "IsReverseComplement" set to "Y" for Index2, then Index2 is processed in reverse complement orientation. Any trimming should be applied to the end of the sequence as it transitions to the adapter, which means the "N" cycles specified in OverrideCycles must be in the beginning of the index read.
Example of 8bp i5 index with the last two (adapter) bases to be trimmed:
Forward i5: XXATCGCGGT
ReverseComp i5: ACCGCGATXX (this is the direction of sequencing)
OverrideCycles for Index2: N2I8
where XX is part of the adapter sequence.
Although Index2 is processed on the sequencer in reverse complement format, Index2 and OverrideCycles are entered in the Sample Sheet in forward, non-complemented format for user convenience (see diagram below).
Note that NovaSeq X has a RunInfo.xml with "IsReverseComplement" set to "Y" for Index2,
FastqCompressionFormat
Optional
The compression format for the FASTQ output files. Allowed values are gzip or dragen.
Sample_ID
Required
ID for the sample with the following requirements: • Must be alphanumeric string with _ or - and no spaces. • Case sensitive (i.e., a Sample Sheet with the samples MySample and mysample is not allowed)
The same Sample_ID may exist on more than one row of the Sample Sheet (e.g., one sample spanning more than one lane)
Undetermined is not allowed as a Sample_ID
Index
Optional
The Index 1 (i7) index sequence. Format is a sequence using ACTG. Required if index cycles are specified for the sample in the OverrideCycles.
Must adhere to the following requirements:
Can only contain A, C, G, or T
Length of string must match number of first index cycles in RunInfo.xml or the number specified in OverrideCycles
A value of na must be used if:
No indexes are specified in OverrideCycles for a sample
Index2
Optional
See description of Index, applied to Index2.
KeepFastQ
Optional
Select whether FASTQs are saved (true) or discarded (false).
Sample_ID
Required
See description in the BCL Convert section
BarcodeMismatchesIndex1
Optional
Specifies barcode mismatch tolerance for Index 1. Possible values are 0, 1, or 2, or na. The default value is 1. Only allowed if Index is specified in RunInfo.xml file and in the Reads section of the Sample Sheet.
A value of na must be used if:
BarcodeMismatchesIndex1 exists in the Data section of the Sample Sheet, and
OverrideCycles exist in the data section, and
Index 1 is masked out for the sample in the OverrideCycles
BarcodeMismatchesIndex2
Optional
See description of BarcodeMismatchesIndex1, applied to BarcodeMismatchesIndex2.
OverrideCycles
Optional
Specifies the sequencing and indexing cycles to be used when processing the sequencing data. Must adhere to the following requirements:
• Must be same number of fields (delimited by
semicolon) as sequencing and indexing reads specified in RunInfo.xml or and in the Reads section of the Sample Sheet.
• Indexing reads are specified with I, sequencing reads are specified with Y, UMI cycles are specified with U, and trimmed
reads are specified with N.
• The number of cycles specified for each read must equal the number of cycles specified for that read in the RunInfo.xml file and in the Reads section of the Sample Sheet.
• Only one Y or I sequence can be specified per read.
'I' cycles can only be specified for index reads
'Y' cycles can only be specified for genomic reads
The total number of cycles used for demultiplexing by both indexes together cannot exceed 27. Note that this includes all cycles between the first "I" cycle used by any sample and the last "I" cycle used by any sample within each index.
"I5N3;I5N3" : counts as 10 bases toward the limit of 27
"I4N1I3;I5N3": counts as (8+5) 13 bases toward the limit of 27
The following are examples of OverrideCycles input: U8Y143;I8;I8;U8Y143 N10Y66;I6;N10Y66
For a sample sheet containing two samples having the following OverrideCycles
Y151; I8N2; N10; Y151
Y151; N2I8; I8N2; Y151
the number of cycles used for demultiplexing sums to 18.
For sequencers with RunInfo.xml having "IsReverseComplement" set to "Y" for Index2, then Index2 is processed in reverse complement orientation. Any trimming should be applied to the end of the sequence as it transitions to the adapter, which means the "N" cycles specified in OverrideCycles must be in the beginning of the index read.
Example of 8bp i5 index with the last two (adapter) bases to be trimmed:
Forward i5: XXATCGCGGT
ReverseComp i5: ACCGCGATXX (this is the direction of sequencing)
OverrideCycles for Index2: N2I8
where XX is part of the adapter sequence.
Although Index2 is processed on the sequencer in reverse complement format, Index2 and OverrideCycles are entered in the Sample Sheet in forward, non-complemented format for user convenience (see diagram below).
Note that NovaSeq X has a RunInfo.xml with "IsReverseComplement" set to "Y" for Index2,
FastqCompressionFormat
Optional
The compression format for the FASTQ output files. Allowed values are gzip or dragen.
Sample_ID
Required
ID for the sample with the following requirements: • Must be alphanumeric string with _ or - and no spaces. • Case sensitive (i.e., a Sample Sheet with the samples MySample and mysample is not allowed)
The same Sample_ID may exist on more than one row of the Sample Sheet (e.g., one sample spanning more than one lane)
Undetermined is not allowed as a Sample_ID
Index
Optional
The Index 1 (i7) index sequence. Format is a sequence using ACTG. Required if index cycles are specified for the sample in the OverrideCycles.
Must adhere to the following requirements:
Can only contain A, C, G, or T
Length of string must match number of first index cycles in RunInfo.xml or the number specified in OverrideCycles
A value of na must be used if:
No indexes are specified in OverrideCycles for a sample
Index2
Optional
See description of Index, applied to Index2.
KeepFastQ
Optional
Select whether FASTQs are saved (true) or discarded (false).
Sample_ID
Required
See description in the BCL Convert section
KeepFastQ
Optional
Select whether FASTQs are saved (true) or discarded (false).
Sample_ID
Required
See description in the BCL Convert section
KeepFastQ
Optional
Select whether FASTQs are saved (true) or discarded (false).
Sample_ID
Required
See description in the BCL Convert section
Bedfile
Required
BED file to be used for analysis in text (*.txt) or gzip (*.gz) format. Alphanumeric string with _ or - or . with no spaces allowed.
KeepFastQ
Optional
Select whether FASTQs are saved (true) or discarded (false).
GermlineOrSomatic
Required
Indicator of pipeline type. Accepted values are germline or somatic.
AuxNoiseBaselineFile
Optional
Alphanumeric string with "_" or "-" or ".". and no spaces in text (*.txt) or gzip (*.gz) format. Applicable only if GermlineOrSomatic is Somatic.
AuxCnvPanelOfNormalsFile
Optional
Alphanumeric string with "_" or "-" or "." and no spaces in text (*.txt) or gzip (*.gz) format.
Sample_ID
Required
See description in the BCL Convert section
Bedfile
Required
BED file to be used for analysis in text (*.txt) or gzip (*.gz) format. Alphanumeric string with _ or - or . with no spaces allowed.
KeepFastQ
Optional
Select whether FASTQs are saved (true) or discarded (false).
GermlineOrSomatic
Required
Indicator of pipeline type. Accepted values are germline or somatic.
AuxNoiseBaselineFile
Optional
Alphanumeric string with "_" or "-" or ".". and no spaces in text (*.txt) or gzip (*.gz) format. Applicable only if GermlineOrSomatic is Somatic.
Sample_ID
Required
See description in the BCL Convert section
Bedfile
Required
BED file to be used for analysis in text (*.txt) or gzip (*.gz) format. Alphanumeric string with _ or - or . with no spaces allowed.
KeepFastQ
Optional
Select whether FASTQs are saved (true) or discarded (false).
GermlineOrSomatic
Required
Indicator of pipeline type. Accepted values are germline or somatic.
AuxNoiseBaselineFile
Optional
Alphanumeric string with "_" or "-" or ".". and no spaces in text (*.txt) or gzip (*.gz) format. Applicable only if GermlineOrSomatic is Somatic.
Sample_ID
Required
See description in the BCL Convert section
Bedfile
Required
BED file to be used for analysis in text (*.txt) or gzip (*.gz) format. Alphanumeric string with _ or - or . with no spaces allowed.
KeepFastQ
Optional
Select whether FASTQs are saved (true) or discarded (false).
GermlineOrSomatic
Required
Indicator of pipeline type. Accepted values are germline or somatic.
Sample_ID
Required
See description in the BCL Convert section
See Comparison1 description.
Comparison4
Optional
See Comparison1 description.
Comparison5
Optional
See Comparison1 description.
RnaGeneAnnotationFile
Optional
Genotype reference file. Alphanumeric string with "_" or "-" or "." with no spaces allowed. If DifferentialExpressionEnable is True., the GTF file must be provided by the user or included with the reference genome.
KeepFastQ
Optional
Select whether FASTQs are saved (true) or discarded (false).
DifferentialExpressionEnable
Optional
Accepted values are true or false.
Sample_ID
Required
See description in the BCL Convert section
Comparison1
Optional
Accepted values are control, comparison, or na. Use only if DifferentialExpressionEnable is true. Must have at least 2 control and 2 comparison samples. May have a max of 15 control or comparison samples.
Comparison2
Optional
See Comparison1 description.
Comparison3
Optional
RnaGeneAnnotationFile
Optional
Genotype reference file. Alphanumeric string with "_" or "-" or "." with no spaces allowed. If DifferentialExpressionEnable is True., the GTF file must be provided by the user or included with the reference genome.
KeepFastQ
Optional
Select whether FASTQs are saved (true) or discarded (false).
DifferentialExpressionEnable
Optional
Accepted values are true or false.
Sample_ID
Required
See description in the BCL Convert section
Comparison1
Optional
Accepted values are control, comparison, or na. Use only if DifferentialExpressionEnable is true. Must have at least 2 control and 2 comparison samples. May have a max of 15 control or comparison samples.
Comparison2
Optional
See Comparison1 description.
Comparison3
Optional
RnaGeneAnnotationFile
Optional
Genotype reference file. Alphanumeric string with "_" or "-" or "." with no spaces allowed. If DifferentialExpressionEnable is True., the GTF file must be provided by the user or included with the reference genome.
KeepFastQ
Optional
Select whether FASTQs are saved (true) or discarded (false).
DifferentialExpressionEnable
Optional
Accepted values are true or false.
Sample_ID
Required
See description in the BCL Convert section
Comparison1
Optional
Accepted values are control, comparison, or na. Use only if DifferentialExpressionEnable is true. Must have at least 2 control and 2 comparison samples. May have a max of 15 control or comparison samples.
Comparison2
Optional
See Comparison1 description.
Comparison3
Optional
RnaGeneAnnotationFile
Optional
Genotype reference file. Alphanumeric string with "_" or "-" or "." with no spaces allowed. If DifferentialExpressionEnable is True., the GTF file must be provided by the user or included with the reference genome.
KeepFastQ
Optional
Select whether FASTQs are saved (true) or discarded (false).
Sample_ID
Required
See description in the BCL Convert section
RnaGeneAnnotationFile
Optional
Genotype reference file. Alphanumeric string with "_" or "-" or "." with no spaces allowed. If DifferentialExpressionEnable is True., the GTF file must be provided by the user or included with the reference genome.
KeepFastQ
Optional
Select whether FASTQs are saved (true) or discarded (false).
BarcodePosition
Required
Text string indicating the location of the bases corresponding to the Barcode within the specified BarcodeRead. Base positions are indexed starting at position zero in the Read. Examples: 0_15, or 0_8+21_29+43_51 if the barcodes are not contiguous.
UmiPosition
Required
Text string indicating the location of the bases corresponding to the UMI within the specified BarcodeRead. Base positions are indexed starting at position zero in the Read. Example: 16_27
BarcodeRead
Required
Text string indicating the location within the sequencing run of the Barcode Read, which contains both the Barcode and the Umi. Readn, where n is from the set { 1, 2 }
BarcodeSequenceList
Required
List of expected barcode sequences
RnaLibraryType
Required
Valid values are StrandedForward, StrandedReverse, Unstranded
Sample_ID
Required
See description in the BCL Convert section
RnaGeneAnnotationFile
Optional
Genotype reference file. Alphanumeric string with "_" or "-" or "." with no spaces allowed. If DifferentialExpressionEnable is True., the GTF file must be provided by the user or included with the reference genome.
KeepFastQ
Optional
Select whether FASTQs are saved (true) or discarded (false).
BarcodePosition
Required
Text string indicating the location of the bases corresponding to the Barcode within the specified BarcodeRead. Base positions are indexed starting at position zero in the Read. Examples: 0_15, or 0_8+21_29+43_51 if the barcodes are not contiguous.
UmiPosition
Required
Text string indicating the location of the bases corresponding to the UMI within the specified BarcodeRead. Base positions are indexed starting at position zero in the Read. Example: 16_27
BarcodeRead
Required
Text string indicating the location within the sequencing run of the Barcode Read, which contains both the Barcode and the Umi. Readn, where n is from the set { 1, 2 }
BarcodeSequenceList
Required
List of expected barcode sequences
RnaLibraryType
Required
Valid values are StrandedForward, StrandedReverse, Unstranded
Sample_ID
Required
See description in the BCL Convert section
RnaGeneAnnotationFile
Optional
Genotype reference file. Alphanumeric string with "_" or "-" or "." with no spaces allowed. If DifferentialExpressionEnable is True., the GTF file must be provided by the user or included with the reference genome.
KeepFastQ
Optional
Select whether FASTQs are saved (true) or discarded (false).
BarcodePosition
Required
Text string indicating the location of the bases corresponding to the Barcode within the specified BarcodeRead. Base positions are indexed starting at position zero in the Read. Examples: 0_15, or 0_8+21_29+43_51 if the barcodes are not contiguous.
UmiPosition
Required
Text string indicating the location of the bases corresponding to the UMI within the specified BarcodeRead. Base positions are indexed starting at position zero in the Read. Example: 16_27
BarcodeRead
Required
Text string indicating the location within the sequencing run of the Barcode Read, which contains both the Barcode and the Umi. Readn, where n is from the set { 1, 2 }
BarcodeSequenceList
Required
List of expected barcode sequences
RnaLibraryType
Required
Valid values are StrandedForward, StrandedReverse, Unstranded
Sample_ID
Required
See description in the BCL Convert section
RnaGeneAnnotationFile
Optional
Genotype reference file. Alphanumeric string with "_" or "-" or "." with no spaces allowed. If DifferentialExpressionEnable is True., the GTF file must be provided by the user or included with the reference genome.
KeepFastQ
Optional
Select whether FASTQs are saved (true) or discarded (false).
BarcodePosition
Required
Text string indicating the location of the bases corresponding to the Barcode within the specified BarcodeRead. Base positions are indexed starting at position zero in the Read. Examples: 0_15, or 0_8+21_29+43_51 if the barcodes are not contiguous.
UmiPosition
Required
Text string indicating the location of the bases corresponding to the UMI within the specified BarcodeRead. Base positions are indexed starting at position zero in the Read. Example: 16_27
BarcodeRead
Required
Text string indicating the location within the sequencing run of the Barcode Read, which contains both the Barcode and the Umi. Readn, where n is from the set { 1, 2 }
BarcodeSequenceWhitelist
Required
List of expected barcode sequences
RnaLibraryType
Required
Valid values are StrandedForward, StrandedReverse, Unstranded
Sample_ID
Required
See description in the BCL Convert section
KeepFastQ
Optional
Select whether FASTQs are saved (true) or discarded (false).
DnaBedFile
Required
BED file to be used for analysis in text (*.txt) or gzip (*.gz) format. Alphanumeric string with _ or - or . with no spaces allowed.
DnaGermlineOrSomatic
Required
Indicator of "germline" or "somatic" type for DNA samples
Sample_ID
Required
See description in the BCL Convert section
DnaOrRna
Required
Indicator of a DNA or RNA sample. Valid values are dna and rna.
KeepFastQ
Optional
Select whether FASTQs are saved (true) or discarded (false).
DnaBedFile
Required
BED file to be used for analysis in text (*.txt) or gzip (*.gz) format. Alphanumeric string with _ or - or . with no spaces allowed.
DnaGermlineOrSomatic
Required
Indicator of "germline" or "somatic" type for DNA samples
Sample_ID
Required
See description in the BCL Convert section
DnaOrRna
Required
Indicator of a DNA or RNA sample. Valid values are dna and rna.
KeepFastQ
Optional
Select whether FASTQs are saved (true) or discarded (false).
DnaBedFile
Required
BED file to be used for analysis in text (*.txt) or gzip (*.gz) format. Alphanumeric string with _ or - or . with no spaces allowed.
DnaGermlineOrSomatic
Required
Indicator of "germline" or "somatic" type for DNA samples
Sample_ID
Required
See description in the BCL Convert section
DnaOrRna
Required
Indicator of a DNA or RNA sample. Valid values are dna and rna.
KeepFastq
Optional
Select whether FASTQs are saved (true) or discarded (false).
Sample_ID
Required
See description in the BCL Convert section
See Comparison1 description.
See Comparison1 description.
See Comparison1 description.




















